金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
登陆有奖并可浏览互动!
您需要 登录 才可以下载或查看,没有账号?立即注册


×

点击蓝字↑↑↑“微生态”,轻松关注不迷路
https://www.bioincloud.tech/ (二维码自动识别)
生科云网址:https://www.bioincloud.tech
<hr/>编译:微科盟杨滢,编辑:微科盟居居、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读
过去二十年来,从海洋系统中检索到的微生物基因组数量显著增加。然而,将海洋基因组多样性应用于生物技术和生物医学领域仍然具有挑战性。作者从公开的海洋宏基因组中回收了43191个细菌和古细菌基因组,具有广泛的多样性,包含138个不同门,重新定义了海洋细菌基因组大小的上限,并揭示了CRISPR-Cas系统和抗生素抗性基因之间的复杂联系。对这些海洋基因组进行计算机生物勘探,发现了一种新的CRISPR-Cas9系统,10种抗菌肽和3种降解聚对苯二甲酸乙二醇酯的酶。通过体外实验证实了其有效性和功效。这项研究表明全球规模的测序计划促进了人们对微生物多样性如何在海洋中进化和维持的理解,并证实如何可持续地利用这些计划来推进生物技术和生物医学。
论文ID
原名:Global marine microbial diversity and its potential in bioprospecting
译名:全球海洋微生物多样性及其在生物勘探中的潜力
期刊:Nature
IF:50.5
发表时间:2024.9
通讯作者:章文蔚、范广益、孙颖、李盛英、Thomas Mock
通讯作者单位:华大生命科学研究院、山东大学、英国东安格利亚大学
DOI号:10.1038/s41586-024-07891-2
实验设计
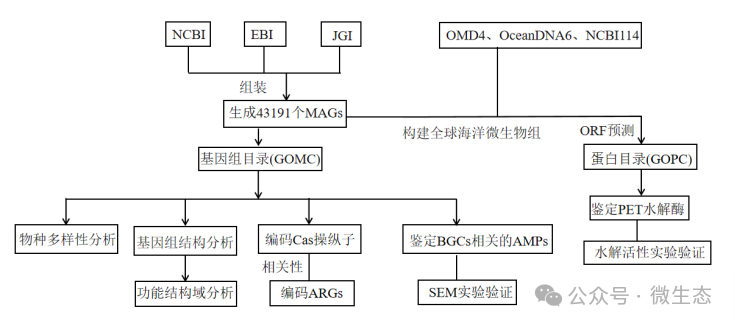
结果
海洋中细菌和古细菌约10
29 个,是支撑全球生物地球化学流动和生态过程的重要组成部分,具有广泛的分类和代谢多样性,并能迅速适应环境变化。随着测序技术的发展,已经克服了不可培养性带来的障碍,从而使基因组水平上的宏基因组学能够揭示海洋生物多样性。特别是一些具有这样意义的项目,如全球海洋采样%28GOS%29和塔拉海洋考察,极大地丰富了研究人员对全球范围内海洋微生物资源库的认识。尽管有这些全球性的测序工作,但只有少数研究使用了一种全面的方法来评估全球海洋微生物群的功能多样性。Nayfach等人也采用了类似的方法,侧重于陆地和宿主相关的微生物组,但没有对预测的生物技术潜力进行实验验证。关于生物技术潜力,已有初步的实验数据表明,海洋微生物群中存在参与氨磷汀和蟒蛇酰胺生物合成途径的基因。因此,需要强有力的实验来评估这些全球微生物组数据集的可用性和价值,以便未来将这些数据集应用于生物技术和生物医学。这一方法需要包含来自各种海洋生态系统的微生物,包括难以评估的极地海洋和深海,以探索巨大的微生物多样性。因此,作者选择了两步走的方法来解决目前的差距:%281%29基于宏基因组学生成一个涵盖所有主要海洋生态系统%28包括极地海洋和深海%29的全面统一目录。%282%29将生物信息学与实验方法相结合,提供有力证据证明海洋微生物组是海洋生物勘探的宝贵资源。
为了实现这一目的,为海洋生物勘探奠定基础,作者分析了2009年8月至2020年7月期间国家生物技术信息中心%28NCBI%29、欧洲生物信息学研究所%28EBI%29和联合基因组研究所%28JGI%29公开提供的海洋宏基因组数据,在3470个微生物属和138门中生成了43191个宏基因组组装基因组%28MAGs%29。结合NCBI、海洋微生物组数据库%28Ocean Microbiomics Database, OMD%29和OceanDNA中公开的海洋细菌和古细菌基因组数据,构建了统一的全球海洋微生物组基因组目录%28GOMC%29。GOMC极大丰富了已知的海洋微生物多样性,在不同的分类等级中有许多新的MAGs。通过分析细菌和古细菌MAGs的丰度,确定了全球范围内微生物组的生物地理模式。通过大量基因组综合统计分析,研究揭示了基因组中编码的微生物适应特征,例如基因组大小和对CRISPR-Cas或抗生素抗性基因%28ARG%29防御系统的偏好。作者还发现了一种新的CRISPR-Cas9系统,几种抗菌肽%28AMPs%29和高活性的嗜盐酶,它们可以降解塑料,并在实验室中证实其活性。基因组目录作为未来研究的宝贵资源,不仅推进研究人员对全球微生物多样性的理解,而且有利于研究如何可持续地利用这种多样性,以减轻环境污染,并通过推进生物技术和生物医学应用造福人类。
1.全球海洋微生物群的扩展
作者从24,395个公开的海洋宏基因组中收集了237.02 Tb的序列数据,涵盖了广泛的海洋环境,从极点到极点%28纬度范围从77.90°S到89.99°N%29,从海洋表面到海沟。从这些宏基因组中组装了43,191个中到高质量的MAGs,平均完整度为82.33%,污染度为1.79%%28图1a,b%29。根据基因组分类数据库%28GTDB%29,共有26、79、304、1185和5783个MAGs无法在门、纲、目、科和属水平上归类。在物种水平上,细菌和古生菌的大部分%28分别为43.37%和43.89%%29无法归属于任何已知的分类单元,占20,295个MAGs。为了提供详细的海洋微生物基因组目录,作者进一步整合了来自三个额外数据库的海洋微生物基因组,包括OMD、OceanDNA和NCBI的8050个公共基因组。得到一个包含24,195个基因组的非冗余目录,构成了GOMC。本研究共组装了9,937个MAGs,占GOMC的41.07%,其中大部分%2882.06%%29代表了先前数据库中没有的潜在新物种%28图1c%29。这些特定的MAGs主要来自深海区%283,713个%29、沉积物%281,371个MAGs%29以及宿主相关%281,250个MAGs%29的生态系统。新的MAGs显著增加了已知海洋微生物组的多样性,占泉古菌门和嗜极古菌门基因组的65%%28图1d%29,占弯曲菌门和脱硫菌门基因组的85%以上%28图1d%29。
除了基因组目录之外,作者还进一步探索了该数据库的生物地理意义。先前的研究主要通过扩增子测序研究海洋微生物群落,特别是海洋微生物群落动力学的方面,也有少数使用宏基因组测序。作者使用统一流形逼近与投影%28UMAP%29降维技术来揭示海洋微生物群落中的生物地理模式。分析确定了56个不同的宏基因组区域%28MPs%29 %28ANOSIM检验,R= 0.61, P< 0.01%29。在全球范围内,MPs并不局限于地理上聚集的采样点,而是表现出大规模的生物地理分区。MPs分布缺乏严格的地理限制,这提出了关于海洋连通性在塑造微生物生物地理中的作用的问题。洋流促进了微生物群落远距离的扩散,从而形成了观测到的全球尺度格局,只有这一解释似乎是合理的。MPs主要局限于特定的海洋深度,在相邻的深度边界上少数例外,因此显示出清晰的深度剖面。这种与深度相关的分离表明存在强烈的环境过滤。除了MPs在生态模式方面的作用外,它们还代表了一个框架,用于在更广泛的地理范围内识别基因组特性,而不是区分MPs特征,如对防御系统的分析所示。
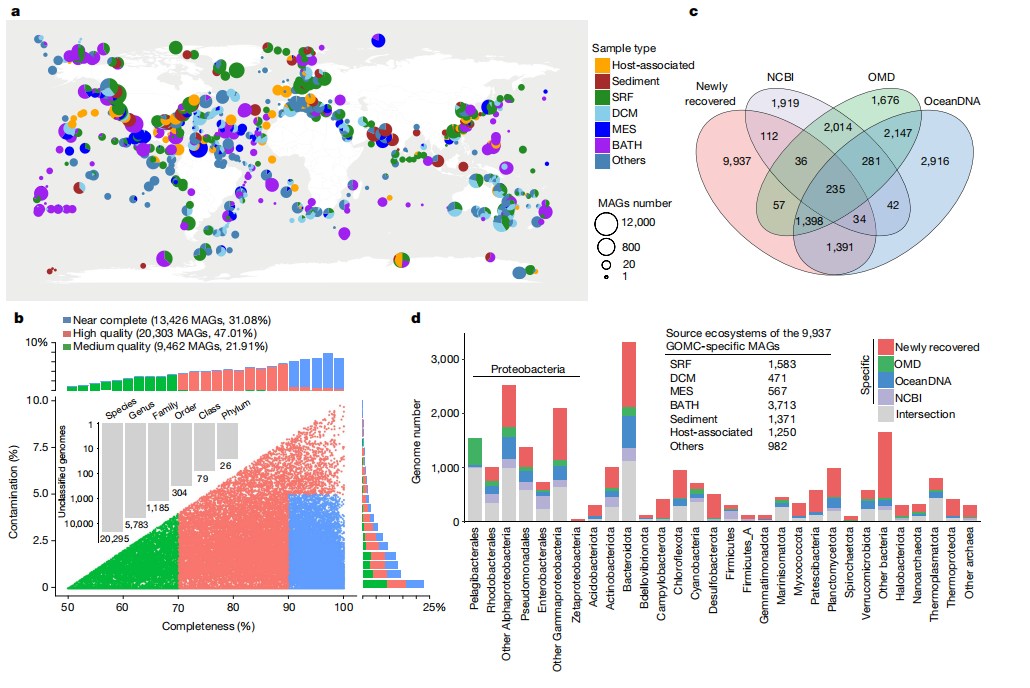
图1.MAGs的地理和生态系统分布。%28a%2943191个新组装的MAGs的地理分布;%28b%29本研究的43,191份中等或更高质量的MAGs构成;%28c%29显示新组装的基因组、NCBI、OMD和OceanDNA之间特有或共有的种水平基因组的维恩图;%28d%29当前研究和现存已发表的数据库对每个细菌和古细菌门的贡献。
2.大型海洋细菌基因组的意义
进化理论预测,高环境变异性会选择具有更高代谢潜力的较大基因组。这在陆地和淡水生态系统中都有记录,但在海洋生态系统中却鲜为人知。在GOMC中,作者发现了303个大基因组,估计基因组大小至少为8 Mb。其中,从浮霉菌门发现的3个基因组大小在16.7 - 18.4 Mb之间的MAGs扩展了已知海洋细菌基因组大小的上限%28图2a%29。这些基因组来自加勒比海委内瑞拉北部大陆架上的缺氧海洋盆地%28Cariaco盆地%29的两个样本。其近缘物种Pirellulaceae,基因组大小为11.7 Mb,发现于黑海缺氧远洋系统的上层。尽管这两种环境在一些理化性质上有所不同,但它们都具有养分供应波动和显著氧化还原的特征。表明这些生态系统中较大的环境可变性可能会施加选择压力,从而对基因组较大的细菌有利。为了进一步研究基因组特征与大小之间的关系作者评估了总体基因组特征的变化。虽然较小的基因组倾向于采用较高的编码密度,但在主要细菌门中,编码密度和基因组大小之间没有一致的趋势。然而,作者发现随着基因组大小的增加,基因长度和基因间区长度也有所增加。同样,较大的基因组往往具有较高的GC含量,最高可达75%左右,这可能是由于内在突变偏差和环境因素的共同作用。
此外,作者研究了功能基因含量与基因组大小之间的趋势,假设更大的基因组优先积累参与基因组稳定性、细胞周期进程、信号转导和基因调控的基因。利用系统发育回归分析,重构了祖先的蛋白质组,并探索基因组大小和基因拷贝之间的关系,最终确定了77个可能有利于基因组大小扩展的Pfam结构域。在较大范围内,这些结构域中的大多数与基因组大小表现出显著的正相关%28图2b%29和广泛的功能作用,如营养获取,对环境刺激的反应以及与其他生物的相互作用。例如,甲基转移酶结构域%28PF08241和PF13649%29可能是基因组大小的重要预测因子。已有研究证明细菌DNA甲基化在基因调控、基因组稳定性和防御机制中发挥作用。因此,具有更大基因组的细菌可能编码更多样化的基因和调控元件,从而增加了DNA甲基化模式的复杂性。这些生物也可能具有复杂的防御系统,利用DNA甲基化来抵御噬菌体感染和外来DNA。血管性血友病因子A型结构域%28PF13519%29是影响细菌粘附、生物膜形成和细胞相互作用的关键结构基本单元,这是另一个值得注意的指标。它的模块化特性促进了关键的蛋白质-蛋白质相互作用,这对细菌在不同环境下的粘附至关重要,并且有助于配体识别,影响细菌定植和群落动态。此外,观察到基因组大小与WD40基序蛋白%28PF00400%29之间存在显著的正相关。WD40是一个古老的蛋白结构域家族,最初在真核生物中发现,随后在细菌中发现,特别是在那些表型更为复杂的细菌中。具有WD40基序的蛋白质通常作为蛋白质-蛋白质相互作用的支架,在浮霉菌独特的胞质内膜的形成中具有潜在作用,从而促进真核样的细胞区室分隔。
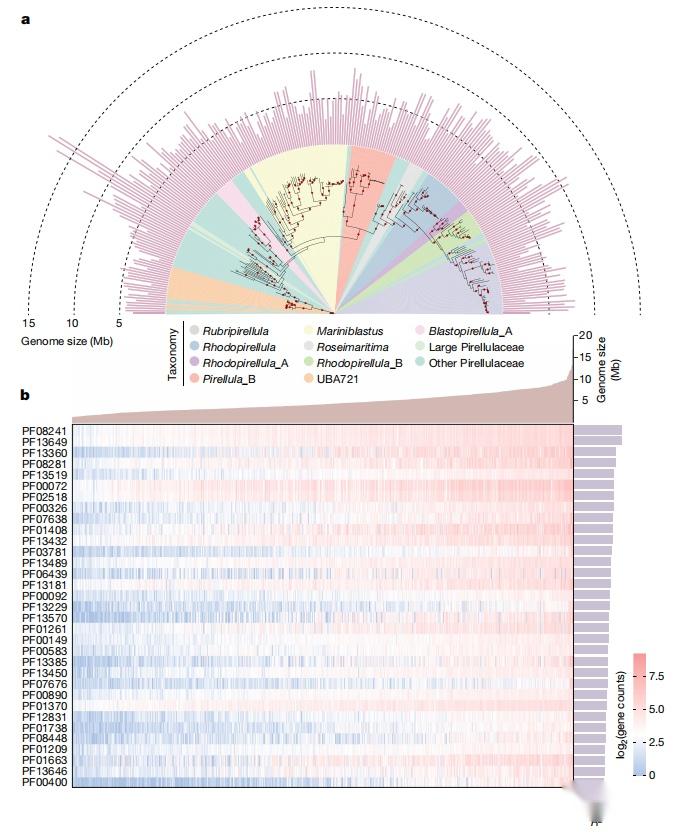
图2.浮霉菌门基因组的大小和功能结构域变异。%28a%29浮霉菌门Pirellulaceae的系统发育树;%28b%29热图显示了浮霉菌门基因组中前33个功能域的分布。每一行对应一个不同的Pfam结构域,每一列代表一个单独的基因组。
3.CRISPR-Cas和ARG系统之间的关系
CRISPR-Cas系统作为一种微生物“免疫”系统,对于防止异质核入侵至关重要。CRISPR-Cas系统在微生物系统发育和不同生态系统中的分布引起了极大关注。在GOMC中,作者从3,212个MAGs中鉴定出40种类型的5,127个Cas操纵子%28约占15%%29,其中1,708个包含完整的CRISPR阵列%28图3a%29。值得注意的是,Firmicutes_B具有编码Cas操纵子的MAGs比例最高,而其他分类群,如Margulisbacteria和Rhizobiales,很少编码Cas蛋白%28图3a%29。这些结果与先前关于Cas操纵子总体存在和跨系统发育不均匀分布模式的研究结果一致。为了探究在Cas操纵子存在方面驱动这种分类偏差的潜在因素,作者分析了温度的影响,温度是先前报道的影响细菌和古细菌中CRISPR-Cas系统丰度的变量。作者预测了GOMC中所有基因组的最佳生长温度,发现在大多数门中,编码Cas蛋白的微生物比没有Cas操纵子的微生物表现出明显更高的平均最佳生长温度。同样,编码CRISPR-Cas系统的MAGs的比例在嗜热菌中明显高于嗜冷菌,在热液喷口样本中也高于开放的海水样本%28图3b、c%29。除了温度之外,与开放海洋相比,宿主相关生态系统显示出更高频率的CRISPR-Cas编码MAGs。厌氧生态系统也显示出相对较高的Cas操纵子患病率,包括肠道微生物组、工程废水厌氧微生物组和陆地深层地下生态系统的微生物组%28图3c%29,这可能先前研究所述,与宿主相关条件以及低氧浓度有关。
尽管微生物CRISPR-Cas系统在阻止外来DNA入侵方面发挥了典型作用,但它们对获得适应性性状%28如抗生素抗性%29的潜在影响仍然是一个有趣的研究领域。作者研究了不同谱系中编码Cas操纵子或缺乏Cas操纵子的基因组中ARGs的频率。观察到在一些微生物门中,与不编码Cas的基因组相比,同时编码Cas的基因组中ARGs的频率显著降低。这些菌生活在有利于选择CRISPR-Cas防御系统的环境中,包括热液喷口的热原体菌门和嗜极古菌门,以及在厌氧或宿主相关环境中通常观察到的髌骨菌、WOR-3、芽单胞菌门、Marinisomatota和厚壁菌门%28图3d%29。然而,Cas的存在并没有降低其余门中编码ARGs的MAGs的比例%28图3d%29。进一步将每个基因组中Cas操纵子的存在及数量考虑在内进行分析,发现它们的数量似乎限制了基因组可能编码的ARGs数量的上限%28P< 0.001%29。因此,随着Cas操纵子数量的增加,ARGs和可移动遗传元件的数量减少%28图3e%29。值得注意的是,先前一些研究发现在选定的致病菌株中CRISPR-Cas和ARGs之间存在负相关,而与磷霉素和利福平耐药相关的基因据在具有CRISPR-Cas系统的大肠杆菌基因组中更为常见。这些观察结果与图3d一致,表明不存在一致的相关性趋势。来自开放海洋的MPs中编码ARGs的MAGs比例明显更高。相反,在宿主相关的MPs中观察到更高比例的编码CRISPR-Cas免疫系统的MAGs,表明与开放海洋环境相比,在这些环境中对外源DNA的保护可能更重要。在与各种海洋生态系统相关的大多数MPs中,可以观察到编码两种免疫系统的MAGs,其频率相对较低。
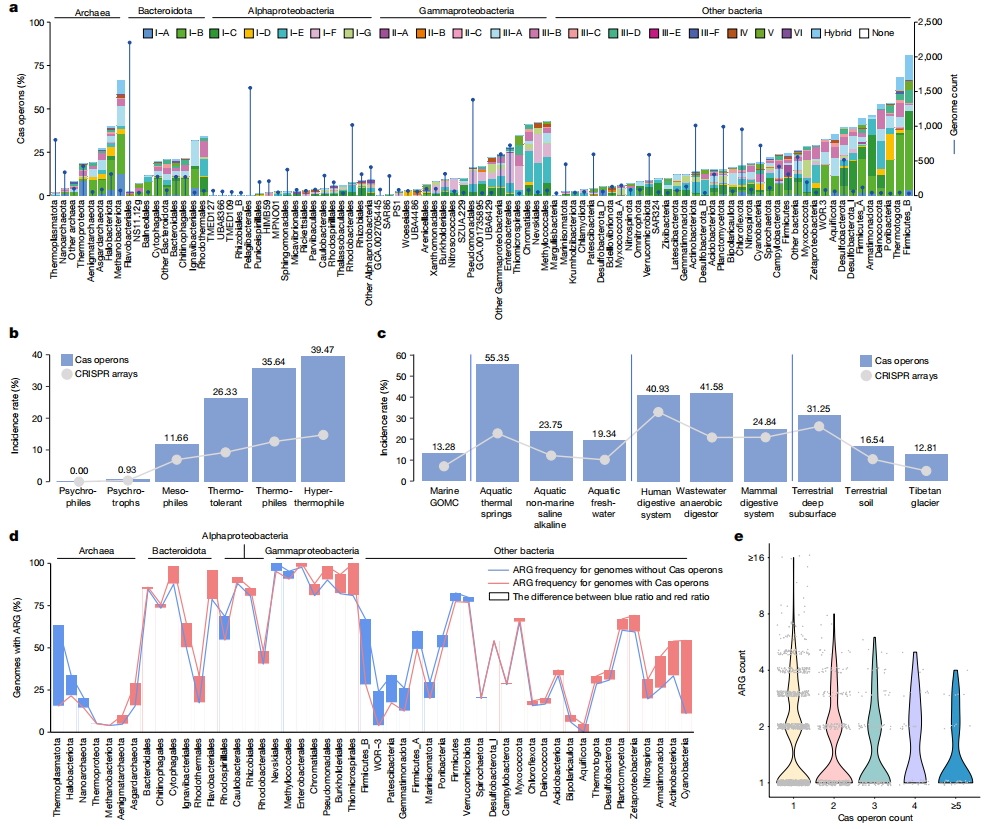
图3.防御系统的分布。%28a%29所有GOMC基因组不同谱系中Cas操纵子的频率;%28b%29不同最佳生长温度下GOMC基因组中Cas操纵子的发生率;%28c%29不同生态系统基因组中Cas操纵子的发生率;%28d%29编码ARG的基因组中有或没有Cas操纵子的部分;%28e%29随着Cas操作子数量的增加,ARG的上限数量呈下降趋势。
4.具有强大体外活性的CRISPR-Cas9系统
已经证明GOMC数据库作为探索新型基因组编辑工具具有宝贵资源的潜力。以应用最广泛的Cas9系统为例,作者鉴定了88个包含Cas9操作子的contigs和完整的CRISPR阵列,其中36个的Cas9蛋白的大小超过950个氨基酸。从中选择了最短的一个%28海洋微生物组CRISPR - Cas9系统%28Om1Cas9%29;1054个氨基酸%29,用于实验检测。Om1Cas9利用由37-bp成熟CRISPR RNA %28crRNA%29和72-bp反式激活crRNA %28tracrRNA%29组成的引导RNA支架,特异性识别3 %27 NNGG原间隔器相邻基序%28PAM%29序列,用于靶向双链DNA %28dsDNA%29。通过在22 ~ 42℃的温度下培养Om1Cas9核糖核蛋白复合物和dsDNA底物进行消化实验。结果表明,Om1Cas9可以在测试的温度范围内有效地切割dsDNA,显示出强大的体外编辑性能。此外,将Om1Cas9序列整合到pX458质粒中,并使用人类细胞评估其活性。具体而言,在血红蛋白亚单位γ %28HBG%29基因和BCL11a增强子区域中选择了5个靶点,并设计了相应的带有合适PAMs的引导RNA间隔物,研究Om1Cas9在治疗β-地中海贫血中的实际应用。在胚胎肾细胞衍生的HEK293T细胞系中,Om1Cas9在HBG和BCL11a增强子基因位点上的切割效率分别为17.08-37.44%和14.89-93.83%。因此,证明了其有效性,并强调了利用GOMC资源鉴定用于各种生物技术应用的新型CRISPR-Cas系统的潜力。
5.对一系列病原体有效的抗菌肽
海洋微生物群落具有合成次生代谢物的能力,具有重要的生态、生物技术和治疗应用潜力。这些分子由生物合成基因簇%28BGCs%29编码。在本研究中,共预测了66种不同类型的64,217个BGCs,长度范围从1,001到576,743 bp。为了解决单个BGCs固有的冗余性和不完整性,将所有BGCs聚类为13063个基因簇家族%28GCFs%29%28图4a%29。值得注意的是,来自5,793个GCFs的约25.49%%2816,369个BGCs%29的BGCs与BiG-FAM参考数据库中任何注释的GCFs仅具有远程相似。大约60.83%的新BGCs被新组装的MAGs特异性编码。大多数新的BGCs来自变形菌门%2838.36%%29和拟杆菌门%2810.65%%29,核糖体合成和翻译后修饰肽%28RiPPs%29%2843.12%%29和萜烯%2823.12%%29是最主要的类型。此外,共鉴定出419个古细菌GCFs,其中233个古细菌特异性结构域主要来自热原体菌门和嗜极古菌门。变形菌门、放线菌门和厚壁菌门的GCFs多样性最高,超过80%的GCFs是门特异性的,这意味着某些次生代谢物的生物合成可能局限于特定的分类群。此外,作者推断了GCFs编码潜力在不同分类等级中的趋势,确定了变形菌门、放线菌门、拟杆菌门和浮霉菌门产生次级代谢物的潜力最高,并强调了属水平分类在评估编码潜力方面的有效性,进一步证实了先前的发现。
作者进行了广泛的数据挖掘,从假设的BGCs中识别潜在的新型AMPs,这些BGCs通常具有各种抗菌和抗肿瘤活性。从629个BGCs中鉴定出1079个推测的AMPs,其中使用深度学习模型从115个BGCs中鉴定出121个独特的候选AMPs %28cAMPs%29。cAMPs主要来自放线菌门%2831个%29、厚壁菌门%2827个%29和变形菌门%2821个%29的羊毛硫多肽II类和I类BGCs。在121个候选AMPs中,117个表现出很高的新cAMPs潜力,这表明海洋微生物群中有丰富的未开发的AMPs来源。
为了验证和表征它们的抗菌活性,通过固态肽合成成功合成了63个少于50个氨基酸的cAMPs。检测它们对5种细菌的抗菌活性,包括革兰氏阳性金黄色葡萄球菌%28ATCC 12600%29和枯草芽孢杆菌%28ATCC 6051%29,以及革兰氏阴性大肠杆菌%28ATCC 25922%29、肺炎克雷伯菌%28ATCC 13883%29和创伤弧菌%28ATCC 27562%29。初步鉴定出10个具有抗菌活性的cAMPs,它们至少抑制了一种菌株的生长。值得注意的是,其中一个被测试的cAMP%28cAMP_87%29对金黄色葡萄球菌和枯草芽孢杆菌的最低抑制浓度%28MIC%29和最低杀菌浓度%28MBC%29为4 μM,而对其他三种菌株,MIC为16 μM, MBC低于32 μM。从盐杆菌科的一种新细菌中初步鉴定出22个氨基酸的肽cAMP_87。AlphaFold2预测的结构表明,cAMP_87采用α -螺旋构象,与AMPs39的典型结构一致。扫描电子显微镜%28SEM%29和透射电子显微镜%28TEM%29图像显示暴露于cAMP_87后细菌膜出现损伤%28图4b%29。因此,cAMP_87对革兰氏阴性菌和革兰氏阳性菌均表现出广谱和有效的抗菌活性。结果表明,新的海洋细菌基因组具有巨大的AMP挖掘潜力,作为海洋微生物基因组尚未开发的新型抗生素空间。
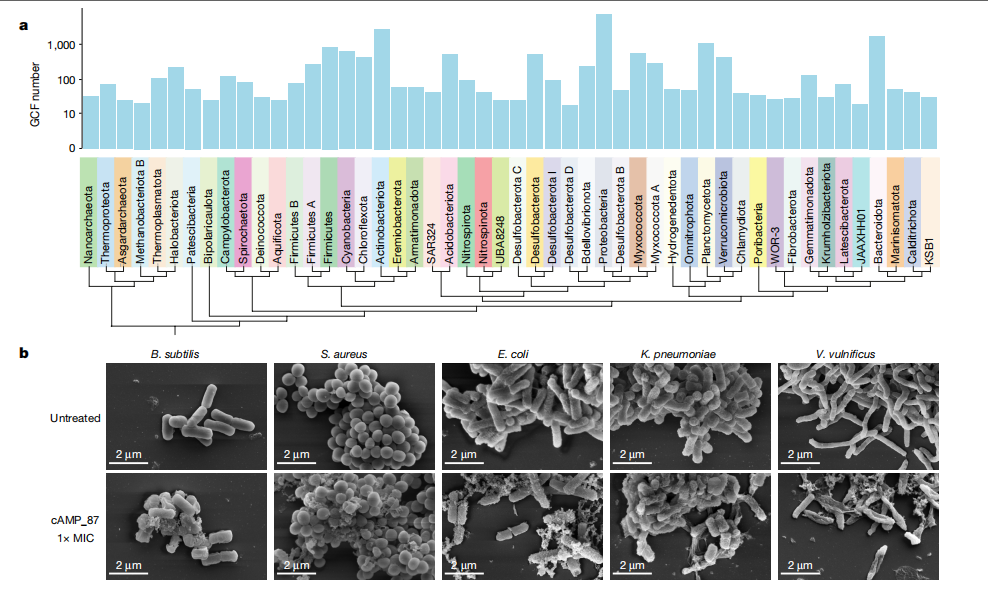
图4.生物合成基因簇和AMPs的鉴定。%28a%29门间生物合成基因簇的比较;%28b%29对5株经cAMP_87处理的菌株和非AMP阴性对照组进行SEM检测,发现细胞内容物渗漏,细胞壁和细胞膜破坏。
6.深海PET酶解聚PET膜
作者通过预测组装的开放阅读框%28ORFs%29构建了全球海洋微生物组蛋白目录%28GOPC%29。GOPC包含超过24.58亿个独特基因,超过海洋微生物参考基因目录40 %28OM-RGC_v2%29的基因数量,为各种生物技术应用的新酶挖掘提供了更全面的资源。自从从一种吸收PET的菌株中发现了一种新的PET水解酶%28IsPETase%29以来,聚对苯二甲酸乙二醇酯%28PET%29的酶解引起了越来越多的关注。定向进化等技术显著提高了PET降解和回收的催化效率。作者使用IsPETase序列作为参考,对GOPC进行了靶向搜索,以发现新的PET水解酶。从不同的海洋生态系统中鉴定出1598个IsPETase同源序列,其中包含保守的Ser-Asp-His催化三元组。这些序列显示出显著的系统发育多样性,形成了不同的进化支,而不受其地理起源的限制。为了鉴定在不同条件下具有良好性能和酶稳定性的PET水解酶,重点研究了与极端海洋环境相关的候选PET酶47。从深沟和热液喷口中分别选择了3个序列,在大肠杆菌中进行了异源表达,并进行了体外生化表征。
以商用GfPET薄膜%28ES301445, Goodfellow%29为底物,检测6种异源表达的深海petase %28dsPETases%29的水解活性。主要水解产物,包括单%282-羟乙基%29对苯二甲酸%28MHET%29和对苯二甲酸%28TPA%29的总浓度%28图5a%29,作为催化活性的一个指标。在6个候选物中,3个亲盐性PET水解酶%28来自北苏热液喷口的dsPETase05,以及来自Mariana海沟的dsPETase01和dsPETase06%29对无定形GfPET膜表现出优异的催化活性,特别是在NaCl浓度升高的情况下%28图5b%29。它们的催化活性随NaCl浓度的增加而增加,在37℃条件下,NaCl浓度为4.5 M或5.3 M时达到峰值。与IsPETase相比,这三种嗜盐PET水解酶的活性分别高出12.0倍、16.0倍和5.6倍%28图5b%29。然而,在不同的盐条件下,dsPETase02、dsPETase03和dsPETase04没有观察到显著的催化活性。喷口流出水生成的dsPETase05的最适温度%2855℃%29高于海沟水生成的dsPETase01和dsPETase06的最适温度%2840和45℃%29%28图5c%29。与不耐盐的IsPETase相比,在最适盐和温度条件下,三种嗜盐的dspetase的活性高出11.8- 44.3倍%28图5c%29。使用最活跃的dsPETase05进行孵育实验,以IsPETase为对照,可视化PET解聚过程。该培养体系包含实验室制备的平均厚度为28 μm的溶剂铸造PET %28scPET%29薄膜和浓度为300和500 nM的petase。在3天的孵育过程中,dsPETase05对scPET膜的可见降解比IsPETase更为显著%28图5d%29。值得注意的是,500 nM的dsPETase05孵育3天后,所有的scPET都被降解成小片段。dsPETse05的解聚率为83%,显著高于IsPETase的41%。同样,当酶的还原浓度为300 nM时,dsPETase05的解聚率仍然高达41%,而IsPETase的解聚率为27%。
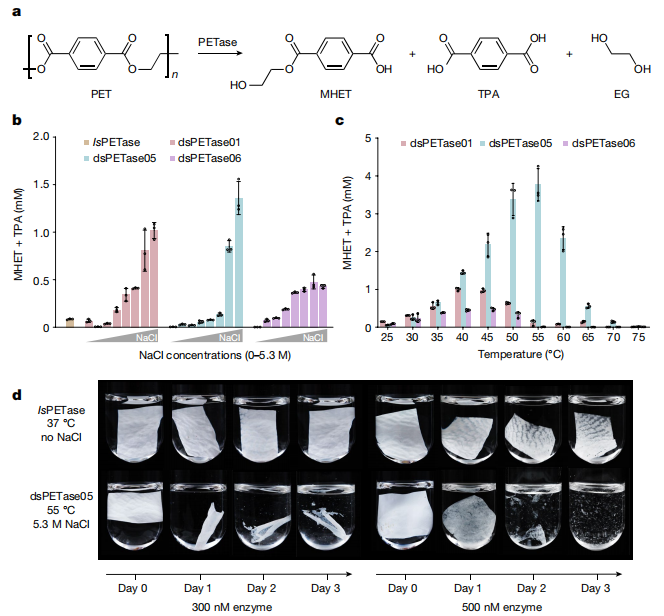
图5.嗜盐水解酶的水解活性。%28a%29PETase催化PET解聚示意图,主要产生可溶性产物MHET、TPA和乙二醇%28EG%29;%28b%29dsPETases的亲盐性。对无定形GfPET薄膜的水解活性由总释放产物浓度%28用HPLC分析MHET和TPA的总和%29表示;%28c%29在一定温度范围内,三种亲盐水解酶对GfPET薄膜的水解活性。dsPETase01和dsPETase05的最佳盐浓度为5.3 M NaCl, dsPETase06的最佳盐浓度为4.5 M NaCl;%28d%29在最佳盐水和温度条件下,亲盐性dsPETase05对scPET膜的降解效果。以IsPETase在无nacl Tris-HCl缓冲液%28pH 9.0%29下37℃催化反应为参照。
讨论
本研究对分布在世界各地的海洋宏基因组和海洋微生物基因组进行了广泛的数据收集和分析。为扩展海洋微生物组的知识做出了重要贡献,并建立了包含24,195个物种水平基因组的GOMC数据库。这一综合资源与蛋白质数据库GOPC提供了对海洋环境内在生物多样性的宝贵见解,并为生物勘探奠定基础。尽管以前基于MAGs的研究已经对海洋系统在维持生物多样性方面的作用提供了初步的见解,但本研究扩展了这些发现,并为可持续勘探和开发提供了数据基础。
由于海洋生境的动态变化,微生物与其环境之间的相互作用在海洋生态系统中具有至关重要的意义。盐度、温度波动、光照可用性和从表面到海底压力的显著变化等因素对微生物种群施加了独特的选择压力,塑造了它们的%28共同%29进化。进化过程导致了基因组大小的差异和适应机制%28如防御系统%29的变化。细菌基因组大小的增加与不同功能域的增加有复杂的关联,这些功能域对营养获取、对环境刺激的反应以及与其他生物体的相互作用至关重要。防御系统在不同生态系统中的数量差异和分布不均反映了海洋的竞争性质。CRISPR-Cas系统的存在强调了它们在动态海洋生态系统中塑造微生物生存策略的重要性。尽管模式复杂且有时相互矛盾,但CRISPR-Cas发生与ARGs防御系统之间的相关性表明,适应性免疫与获得新遗传物质之间存在潜在的联系。基因组可塑性不仅有助于某些生态系统中特定谱系的建立,还体现了海洋微生物的丰富多样性,为有效的生物勘探提供机会。
本研究利用海洋微生物基因组库作为基因组挖掘的基本资源。这种方法能够发现遗传工具和新型生物活性化合物。研究提供了CRISPR-Cas9系统、AMPs和塑料降解酶的宝贵信息,展示了海洋环境中微生物群落中编码的多种分子库。例如,在GOMC中新发现的CRISPR-Cas9系统在各个研究和生物技术领域具有巨大的应用潜力。值得注意的是,链霉菌%28Streptomyces%29、小单孢菌%28Micromonospora%29和假单胞菌%28Pseudomonas_E%29等分类群成为生物勘探工作的潜在候选物种,有助于探索新的BGCs和生物活性化合物。BGCs编码潜力的多样性体现了海洋微生物群落广泛的遗传多样性。值得注意的是,经过实验验证的抗菌肽有望成为对抗多种病原体的抗生素。从体外实验中获得的结果可以反过来改进为精确识别AMPs57的深度学习算法。计算预测和实验验证之间的这种协同关系可能会建立一个正反馈循环,每次迭代都会改进和加强两种方法的有效性。此外,此数据库还具有发现新型酶的巨大潜力,例如识别用于塑料降解和废物管理实践的PET酶。总之,基于深度学习的海洋微生物组基因组挖掘与体外验证相结合,为解决抗微生物药物短缺和海洋污染问题带来了巨大希望,强调了海洋微生物组在促进人类福祉和环境可持续性方面的关键作用。
原文链接:https://doi.org/10.1038/s41586-024-07891-2
获取此篇微文原文pdf请扫描下方二维码联系微科盟多组学老师即可。
微文推荐阅读
- 免费领取100篇近两年土壤微生态研究精选文献合集
- 免费领取100篇近两年植物根际微生态研究精选文献合集
- 免费领取100篇近两年微生物地球化学循环研究精选文献合集
- 免费领取100篇近两年胃肠道微生态研究精选文献合
- 免费领取60篇近两年中草药治疗疾病微生态研究精选文献合集
https://u.wechat.com/MDO_lASfQFBINAsbZgKWO4A (二维码自动识别)
获取此文献原文PDF、申请加入学术群,联系您所添加的任一微科盟组学老师即可,如未添加过微科盟组学老师,请联系多组学老师27,无需重复添加。
原文地址:https://zhuanlan.zhihu.com/p/18772246324 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-3-2 06:29
发表于 2025-3-2 06:29
 提升卡
提升卡


