药企选择NGS检测,是为了什么?
PS:这篇仅代表个人观点。如果我俩理解不一样,那么你是对的。欢迎留言区探讨。 ——听说双击屏幕有惊喜!—— 01 “效率” 血检的机会,源自于组织检测在效率上的“不完美”。 说起来很玄乎,但其实很简单: 要么是干更少的活有同样的产出,要么是干同样的活有更高的产出。 在药物的pivotal study(注册临床)中,与NGS检测合作的 “效率“被具象化成了“入组效率”。 更确切的说,是两个指标: 1.“可检测”的患者比例
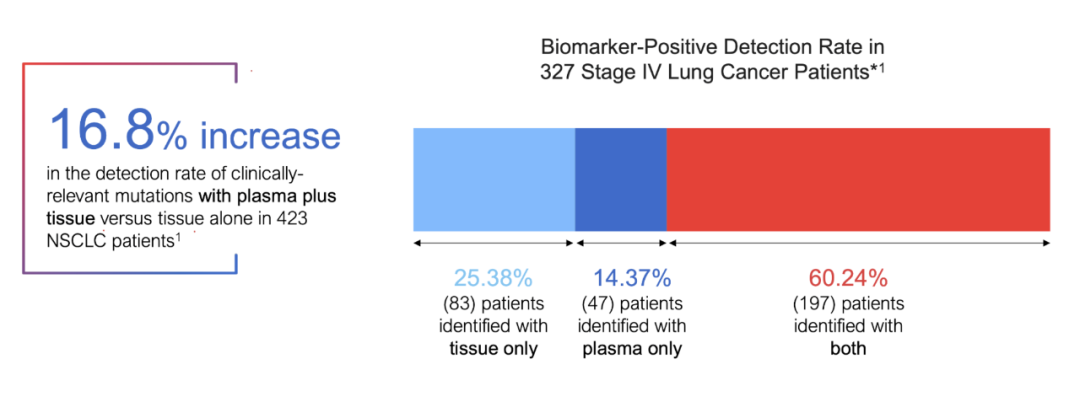
2 .“可入组”的患者比例 “要检测多少患者才能筛选出足够的临床试验入组人数” 在迈过“可检测“这道关卡后,另一道算术题摆在了面前。 在这一点上,客观来说不管是血检only还是组织only,都不是完美的。 2023年,广州第一人民医院的谢教授在BMC Cancer上发表了一篇关于肺癌中组织检测vs液体活检的研究。
研究同时对423例肺癌患者的组织样本和血液样本进行了肿瘤驱动基因(EGFR、ERBB2、ALK、ROS1、C-MET、KRAS、BRAF、RET、BRCA1和BRCA2)检测。 结果显示:
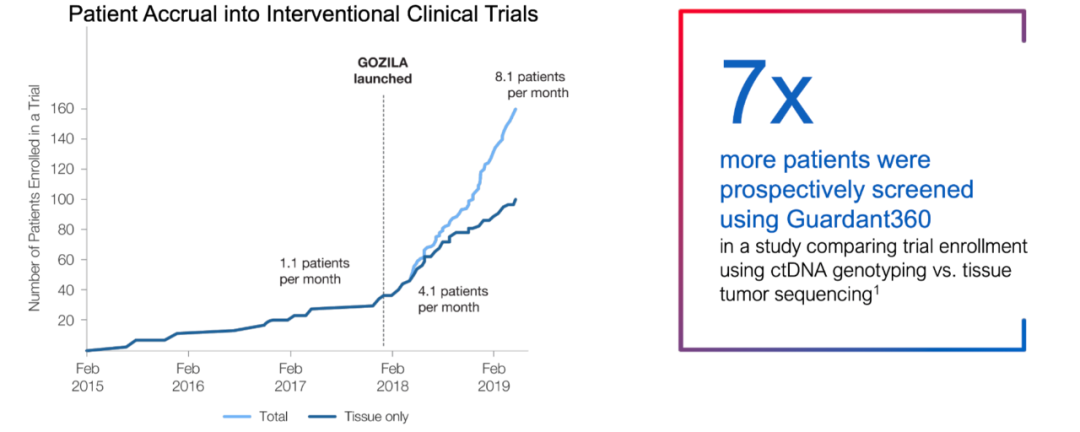
“可检测”的患者比例提升、“可入组“的患者比例提升,在这两个组织检测的效率薄弱点上,NGS血检撕开了药企合作的口子 ——至少,“组织+血液”是一个优于传统组织only的选项。 如果要更为具象化这种优势,发表在Nature Medicine上的GOZILA study是个很好的例子。
02 One “More” Thing
除了与组织检测“相同”的临床试验入组场景外,还额外给出了ctDNA的两个“专属”应用:
1.作为药物开发过程中的疗效指标 2.作为药物临床试验的早期终点 其中有几个观点给血检打开了新的大门: 1. ctDNA水平的变化可用于早期临床试验,以帮助发现药物活性的信号,帮助药企更好的选择药物开发计划。 这种帮助既包括了评估候选药物的抗肿瘤活性,也包括了为后续的临床试验选择最佳的药物剂量。
对于(新)辅助治疗的疗效评估,ctDNA变化或者清零(MRD阳转阴)可能优于传统的基于影像学的评估策略(比如ORR),因为局部治疗的临床试验不能使用基于影像学的判定来衡量患者对治疗的反应。
03 “选择”
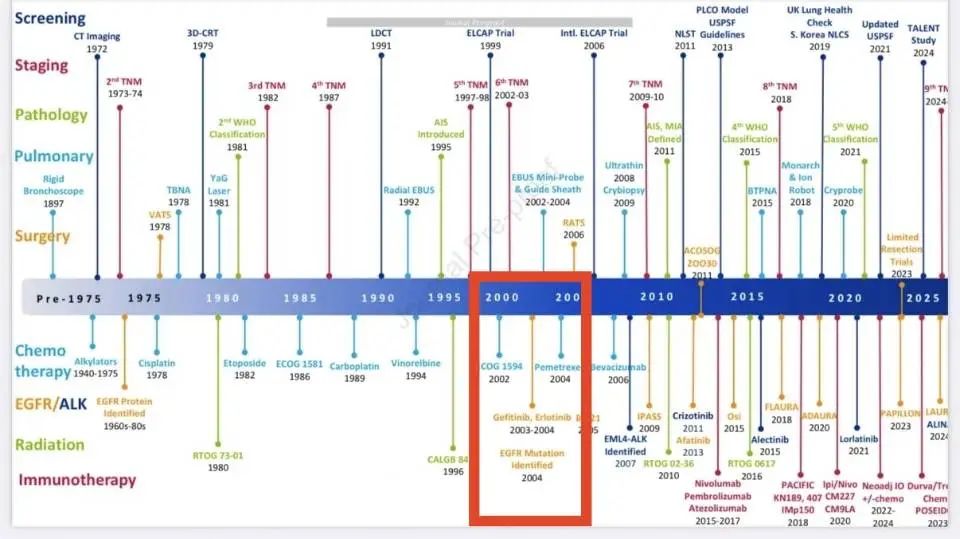
20年前,在吉非替尼的研发过程中,基于组织样本的基因检测发现了EGFR 突变对于药效的巨大影响。 这几乎可以说是“挽救”了这款药物,让其成为了NSCLC的第一款明星靶向药,也开启了肿瘤精准检测的恢弘篇章。 不夸张的讲,那是肿瘤靶向药物研发的“奇点“时刻。 此后的20年,虽然存在很多重大的突破,但组织检测对于药物开发的贡献再也没有迈出EGFR画的那个圈。 20年后,NGS血检有机会引领肿瘤药物研发的下一个“奇点“时刻吗? 监管层给出的信号则更为直接。 2023年1月,FDA批准了Orserdu(艾拉司群)用于ER阳性、HER2阴性、ESR1突变的晚期乳腺癌患者后线治疗。 更重要的是,同步批准了Guardant360 CDx作为其伴随诊断——历史上第一次出现了“liquid-only”的伴随诊断。
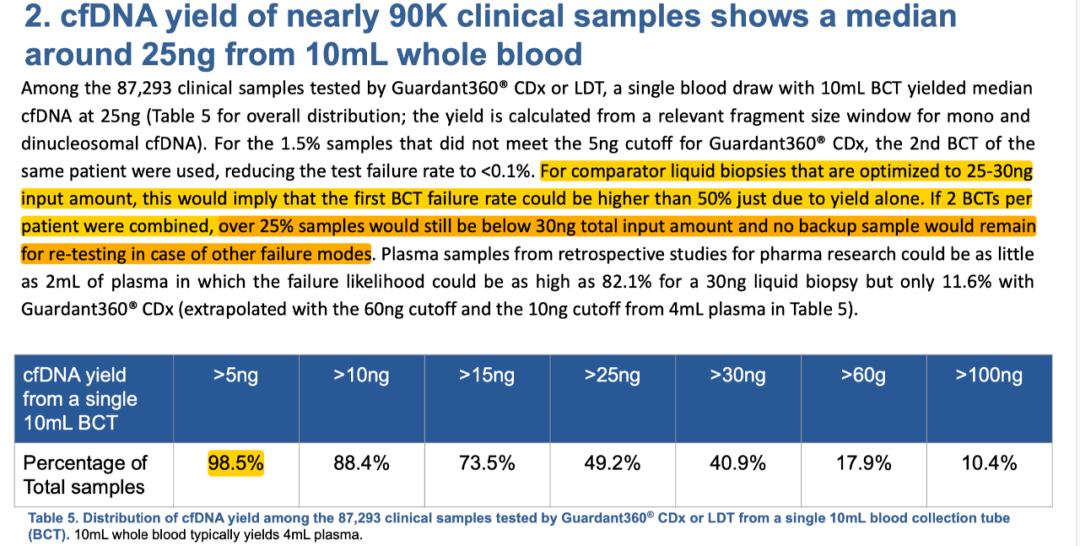
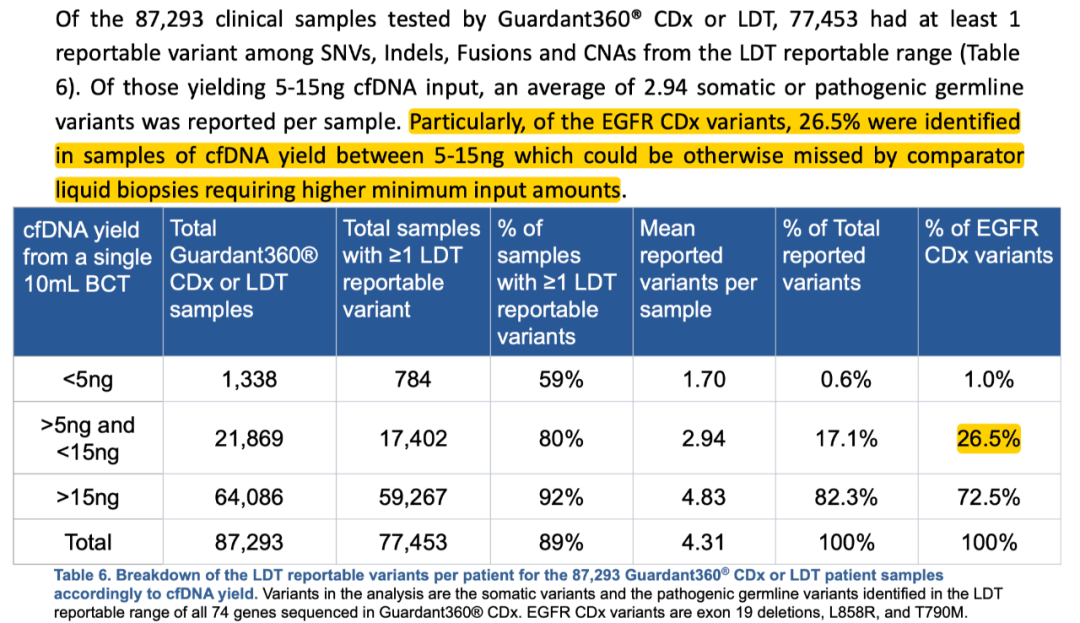
NGS血检已然迎来了其在药企合作上的巨大风口。 04 “壁垒” 相比于组织检测,NGS血检的技术门槛更高、不同厂家之间的技术水平差异更大。 比如一个容易被忽略的指标:DNA投入量。 有的厂家只需要5ng,而有的需要10ng,还有的需要20ng——甚至有些需要30-50ng。 这个看似不起眼的指标会显著的影响我们前面提到的“效率”——也就是“可检测”和“可入组”的患者比例。 在Guardant Health对其检测过的超过8.7万份临床血液样本的回顾性分析中,10ml血液里平均提取的cfDNA中位值只有25ng。 换句话说,只有不到50%的样本(10ml血液)能够提取到超过25ng的cfDNA。 即使将样本量增加一倍——比如用2管血/20ml——依然有超过25%的样本提取的cfDNA总量在30ng以下。
更何况,DNA投入量的需求并不仅仅是“我可以做”这么简单,其背后还包括了是否有足够全面的性能验证、是否有基于不同突变和不同方法学的比较数据、以及这些数据是否具备药物疗效上的可比性 ——这是只有通过长期积累才能构建的数据壁垒,也是超出了单纯价格战的关键选择因素。
|


 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号