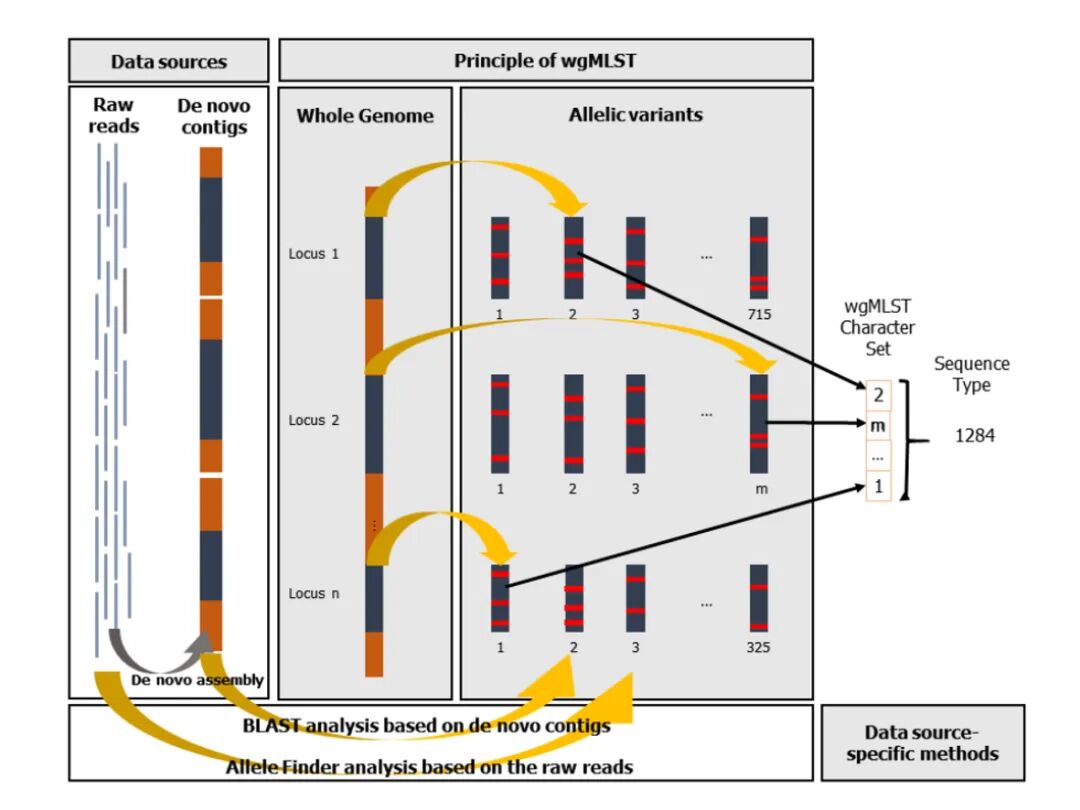
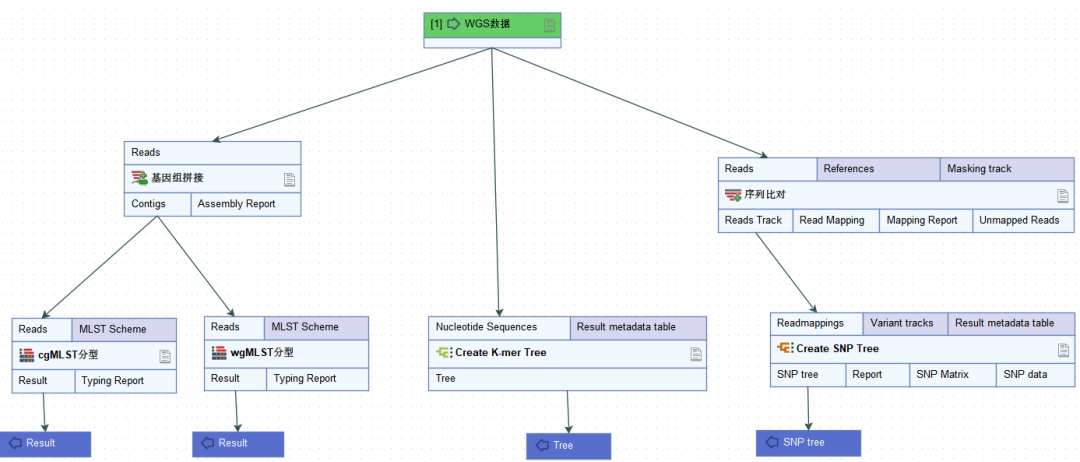
为了分析同一物种内基因组之间的遗传相似性,最初基于7个基因的多位点序列分型方法已扩展到涵盖数百甚至数千个基因位点的核心基因组多位点序列分型(cgMLST)。该方法通过将基因组组装序列与scheme进行比对,从而确定等位基因序列编号。
全基因组多位点序列分型(wgMLST)是cgMLST的进一步延伸,该方法除了使用一组核心基因组位点外,还使用一组辅助基因组位点。实际应用中,通常对cgMLST结果中密切相关的菌株进行wgMLST分析。
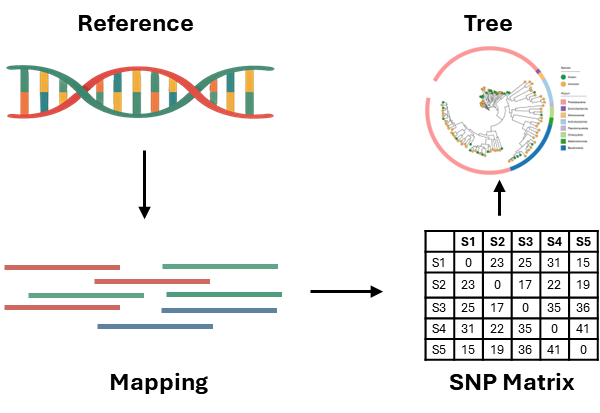
单核苷酸多态性(SNP)是通过将测序数据与最相近的参考序列进行比对,基于核心SNP位点构建进化树,鉴定菌株型别。参考基因组的选择对于SNP分析至关重要,可以采用MLST来确定参考序列。
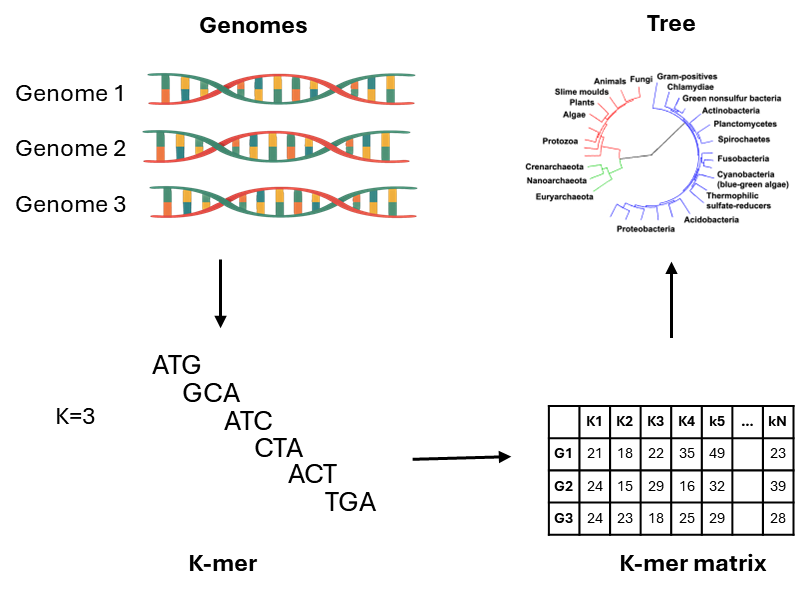
K-mer方法可以克服先验参考基因组和scheme定义的需要。该方法将测序数据分割成特定长度(k)的核苷酸片段。通过比较一组基因组之间K-mer的配对情况,从而评估系统发育相关性。
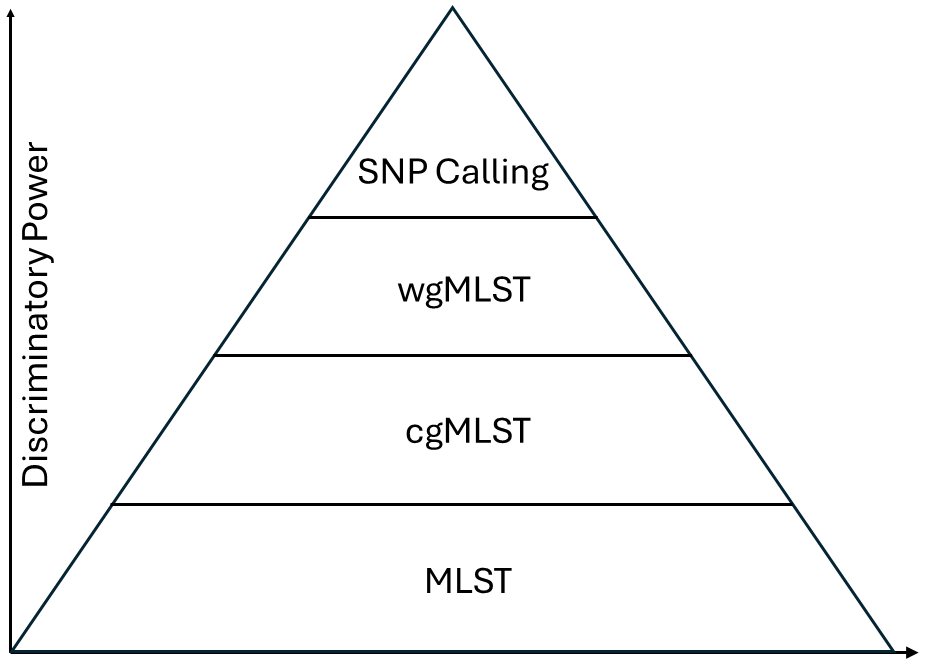
不同分型技术对于微生物鉴定的分辨率从低到高依次为:MLST-> cgMLST -> wgMLST -> SNP Calling。
CLC Genomics Workbench 将不同分型方法整合到一个可视化平台中,能够高效完成从物种鉴定到溯源分型,大大简化了数据分析工作。
参考文献 Uelze L, Grützke J, Borowiak M, Hammerl JA, Juraschek K, Deneke C, Tausch SH, Malorny B. Typing methods based on whole genome sequencing data. One Health Outlook. 2020 Feb 18. |

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号