金桔
金币
威望
贡献
回帖0
精华
在线时间 小时
|
博士的方向涉及一些计算化学,这问题是偶尔看到,占个坑,随时补充修改。
此帖对于我来说 更好像是笔记整理和知识的回顾,如果专门搞量化和模拟的,这些其实应该都很熟悉了,考虑到大家大多是做实验的,我也尽量和实验相结合起来。
我就这些年所学的在计算化学的心得稍微说说。主要包含2010-2020这十年间计算化学的一些发展
要说近十年的发展,就不展开讲更早以前的了,这里就放一个时间表,记录了之前所做的理论方法的重大进步的历史,特别是那些对计算有机化学和生物化学产生影响的理论方法。
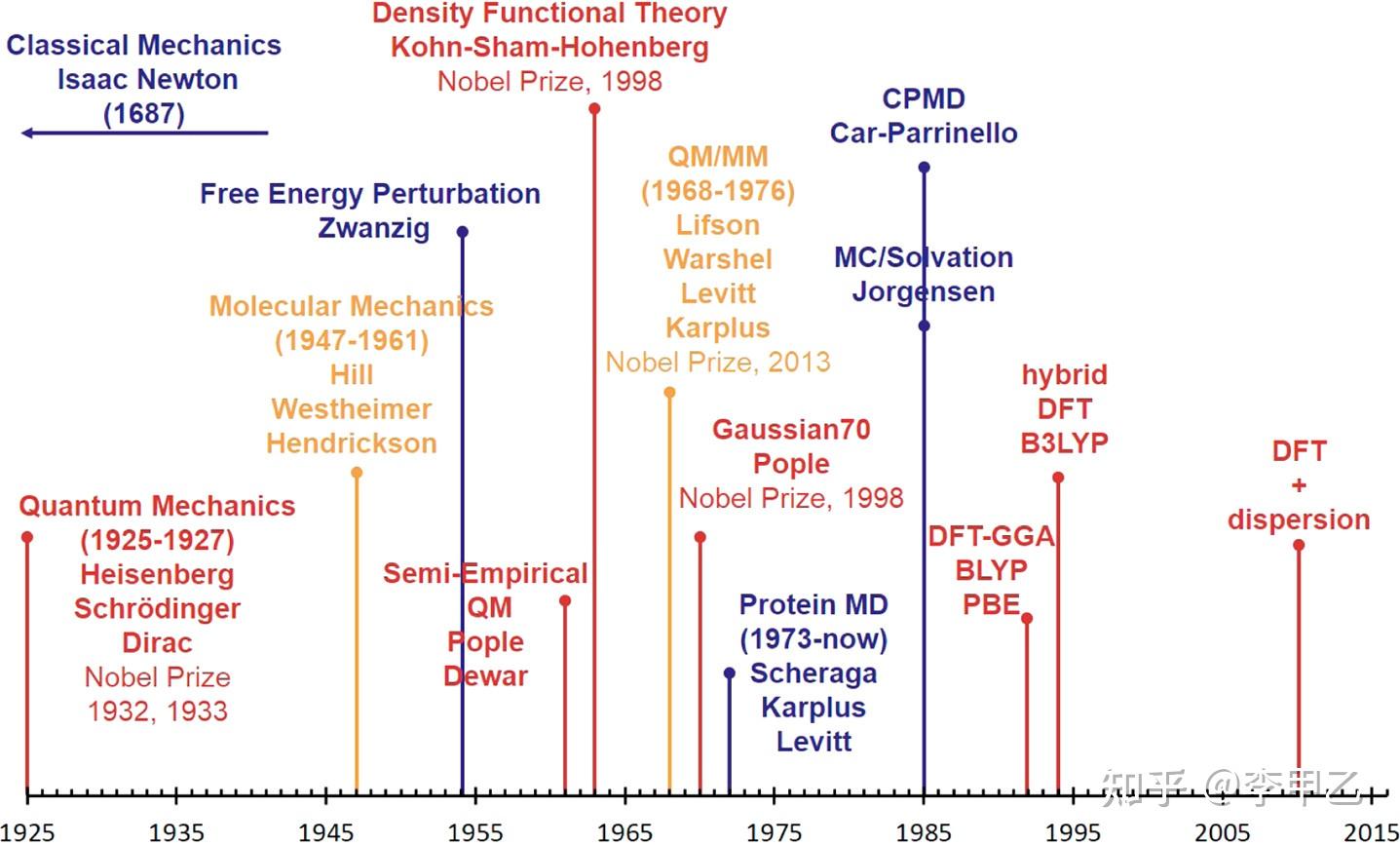
QM / MM组合方法的发展以及密度泛函理论DFT的功能逐步完善,使我们进入了现在的研究领域
然后就讲讲近十年的吧,先说QM这块,再说MD,最后说QM/MM。
1:DFT泛函相关的发展
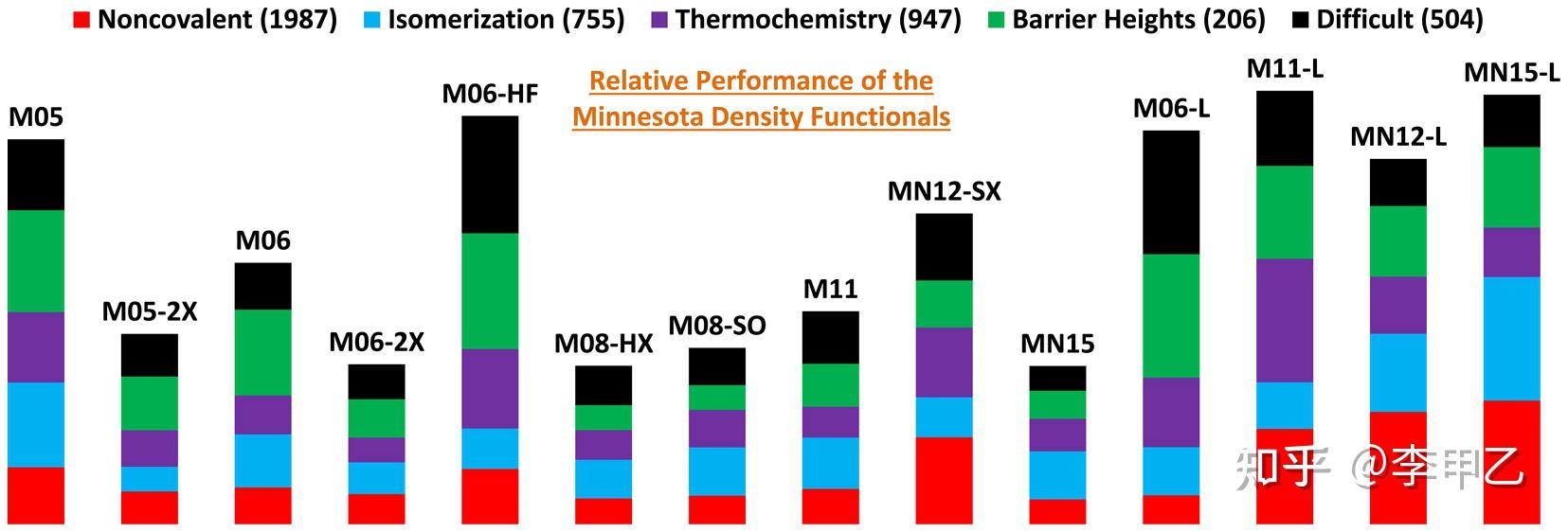
目前日常研究中最常用的泛函是B3LYP,M06-2X,PBE0,wB97XD,PBE,M06,CAM-B3LYP这些。虽然不断有新的泛函提出,但是做量化的大多知道,DFT是存在瓶颈的,虽然实用性不错,精度也挺好,但是继续提升精度没有太多的潜力,基本都是不断去拟合参数,校正修补,和后HF相互杂化来改进。高精度的计算肯定还是后HF,多参考方法的天下,这里贴一张图,来自J. Chem. Theory Comput. 2016, 12, 9, 4303-4325,主要讲了14种明尼苏达州密度泛函的比较,可以发现和最常用的M06-2X相比,后来开发的MN15,MN15-L也并不很突出。
要说明,即便真的找到了精确的交换-相关泛函,形式肯定也很复杂,计算量不会比高级别的后HF小的,所以做实验的同仁或者初学者选用主流常见的DFT泛函即可,多看相关文献心里就有数了。
2:量子化学程序的发展
我是Gaussian的用户,Gaussian发展不大,很多问题,Gaussian16能解决的,其实Gaussian09就可以。但Gaussian还是绝对的主流,每10个量化工作者里就至少有9个人频繁使用Gaussian,其余的ORCA,ADF, Molpro,GAMESS-US,Q-Chem,CP2K,Turbomole等加起来只占一成。
简单说下ORCA
ORCA是免费的,当前是4.1版,优点是支持了很多Gaussian不支持的重要方法,又因为充分利用了RI技术,使得DFT、TDDFT的效率极高,从基态到激发态,从单点到几何优化,从大体系DFT计算到小体系高精度耦合簇计算,以及NEVPT2、MRCI等多参考态计算全都做得挺好。溶剂模型、标量相对论计算、NMR计算等ORCA也都支持。
另外对我平时计算产生便利的是xTB, xTB是做GFN-xTB计算的程序。GFN-xTB(Geometry, Frequency, Noncovalent, eXtended TB),可以以很便宜的方式把几何优化、频率计算、弱相互作用这些问题都算得不错,能用于上千原子大体系。目前最新的是GFN2-xTB,结合了DFT-D4,基本可以做到在计算弱相互作用方面完胜PM7,PM6D3这些。顺手贴个GFN2-xTB的文献链接,引用已近400次了,J. Chem. Theory Comput. 2019, 15, 3, 1652-1671
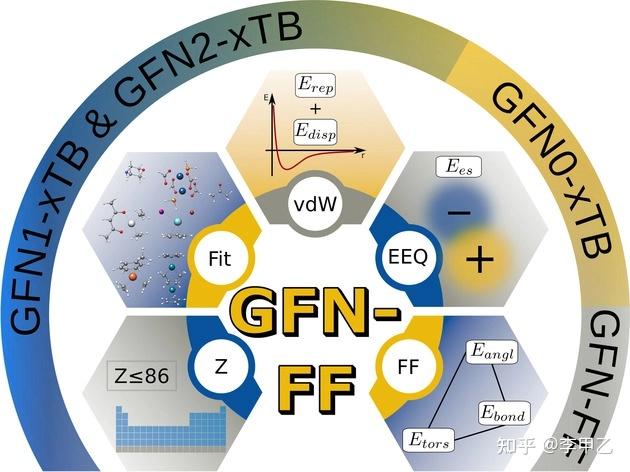
xtb程序目前可以做单点、优化、振动分析等任务,覆盖是元素很全了,体系可以很大,几百上个原子。这是一大优势,多孔金属有机材料,高分子聚酯,都能搞,但是找过渡态、产生IRC,振动分析还做不到。因为大体系算起来很快,做构象搜索的可以利用基于xtb的Crest(Conformer–Rotamer Ensemble Sampling Tool))程序,亲测很方便。
然后计算化学中值得一提的发展和突破不止于此, 不得不提的是DFT-D色散校正的发展
2010年Grimme在JCP上发了篇文章,提出了DFT-D3的思想,2017年他又提出了DFT-D4的思想,做实验的可能不太了解这个的重要性,最直接的我就贴JCP关于DFT-D3文献的被引次数,我记得 知乎上曾经有个问题:有哪些被引过万的文章,喏这篇就是。


我稍微展开说下: 原子在不同化学环境下的色散系数是不同的,比如原子在分脑子中的色散系数就远比它在自由状态小。为了体现这一点,DFT-D3就利用特定公式根据体系坐标计算每个原子的配位数,进而影响色散参数,DFT-D3的本质就是对体系的势能面进行校正,所以单点,几何优化,振动分析,走IRC,AIMD等依赖于势能面的任务都会受到影响。DFT-D3 就可以令描述色散作用不加的泛函都达到了不错的水平,原理上DFT-D3可以结合任何泛函,然后比如结合在B3LYP上就成了B3LYP-D3。 DFT-D3可以用于1-94号元素 。
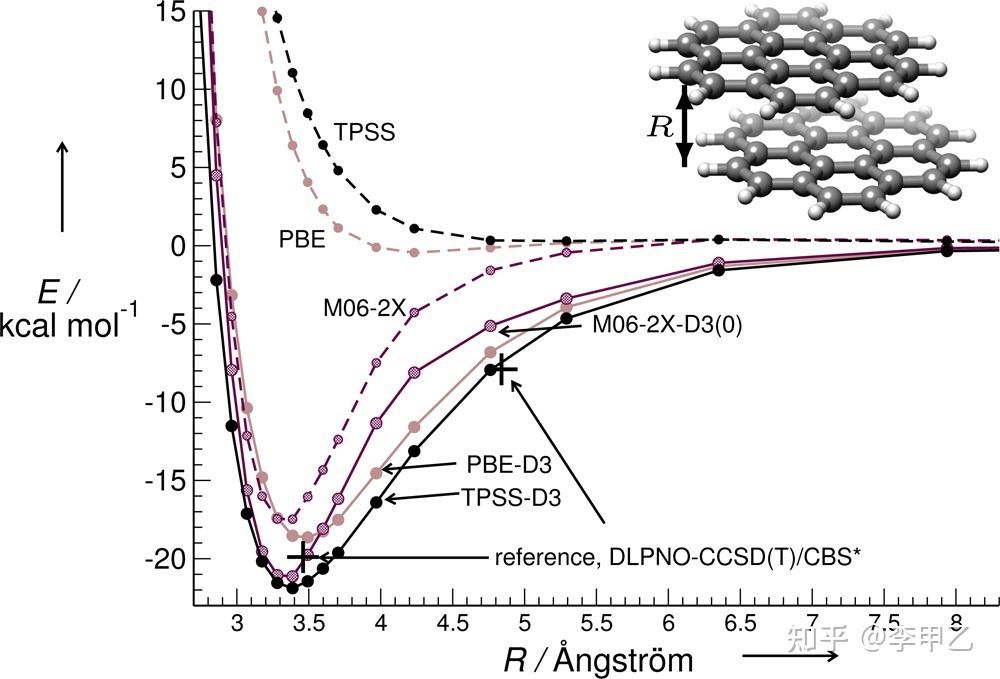
所以体系涉及弱相互作用的,基本都要加色散校正,pai-pai堆积,范德华吸引,氢键作用的存在。或者要研究分子间的结合能,团簇的稳定性,物理吸附,柔性分子构型现象对能量等情况时都会涉及。
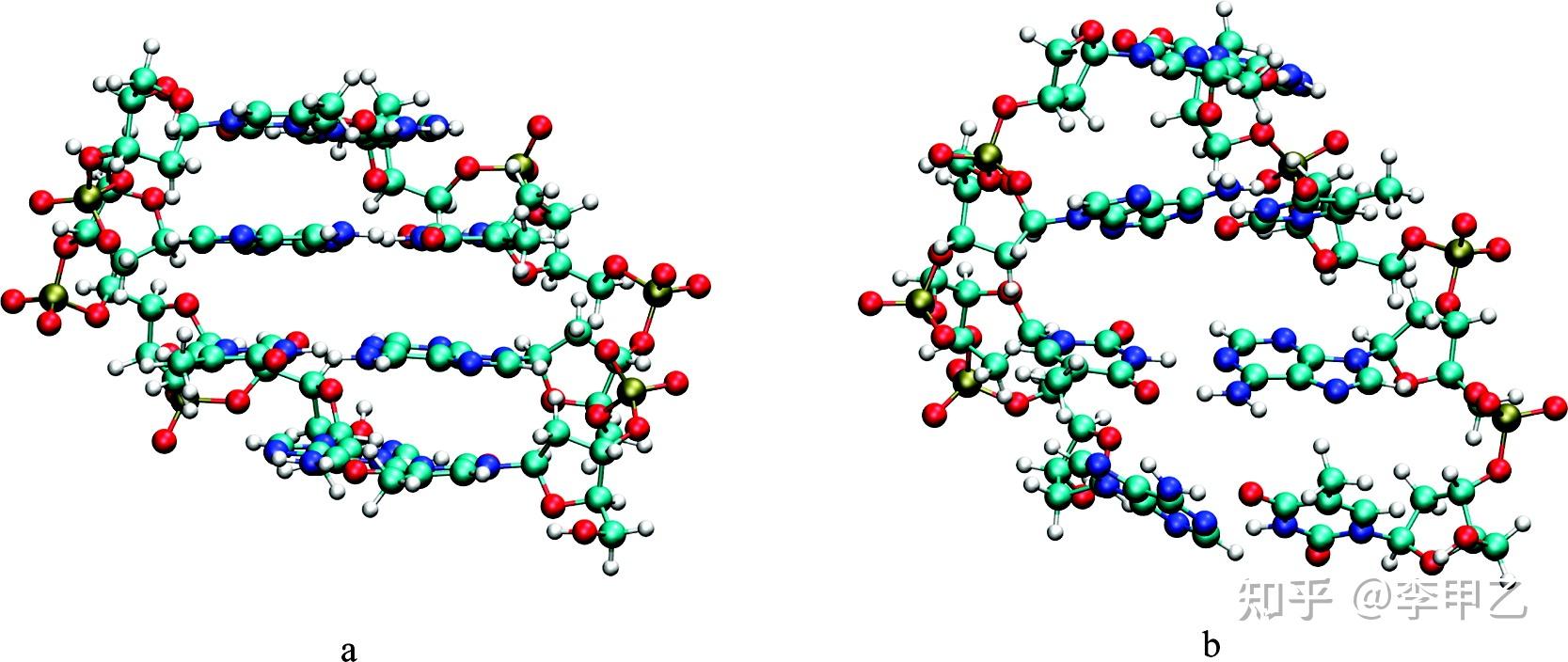
上图来自 J. Am. Chem. Soc. 2008, 130, 47, 16055-16059,左边是加色散校正的,右图没加,导致结构中配对碱基的结构就发送了扭曲,本文的题目也说明了问题:Double-Helica Ladder Structural Transition in the B-DNA is Induced by a Loss of Dispersion Energy.
3:分子动力学(MD)模拟
我主要用AMBER,也会用些Gromacs,没用过CHARMM,NAMD,Lammps。这块发展特别快,我分几个子模块来讲吧:
3.1 力场
蛋白模拟的力场,ff14SB是最主流的,是目前AMBER和Gromacs中可以用的最主流的,这个力场主要是在AMBER99SB-ILDN基础上对骨架参数略微修正,对氨基酸侧链参数重新拟合搞了下,这样就和实验数据对的更准,去年出了个AMBER19SB, 改进了骨架二面角的数据。核酸模拟的话,适合RNA的改进是xOL3,对于DNA的是parmbsc1和OL15,我推荐使用(其实是manual推荐的哈)
OPLS力场的话,最近的改进是OPLS-AA/M 和OPLS-AA/L 但测试表明蛋白质模拟PK不过其它力场
CHARMM力场的话,最近几年是出了个CHARMM36m,对核酸,磷酯,蛋白方面均有改进,比如蛋白是重新拟合了CHARMM22/CMAP的骨架参数,解决其偏向出现α螺旋的倾向,还优化了侧链参数。
溶剂模型的话 顺便提下,最广泛的水模型还是3点水模型(TIP3P和SPC)都是上世纪80年代的事情了,四点水模型TIP4P是OPLS御用的(用TIP3P,SPC也完全可以),OPLS2.0后御用SPC,然后还有五点水,六点水,七点水模型,比较小众,比如七点水唯一的亮点是可以在较大温度范围内较准确地重现密度和介电常数。也有文献提到可极化水模型,因为极性环境会显著对水的电荷分布产生极化作用,但计算量会比固定电荷水模型大很多,且被主流程序支持不广泛,也不是非用不可的,所以我在生物大分子的模拟中也见得不多。对水模型参数的标准优化方法感兴趣的可以看看2014年的一篇JPCL: J. Phys. Chem. Lett. 2014, 5, 11, 1885-1891。
3.2 采样方法
我们知道MD模拟基本就是干两件事,一是采样,也就是构象空间的搜索,二是自由能的计算
先说采样,为什么采样这要紧又这么难呢 且看下图
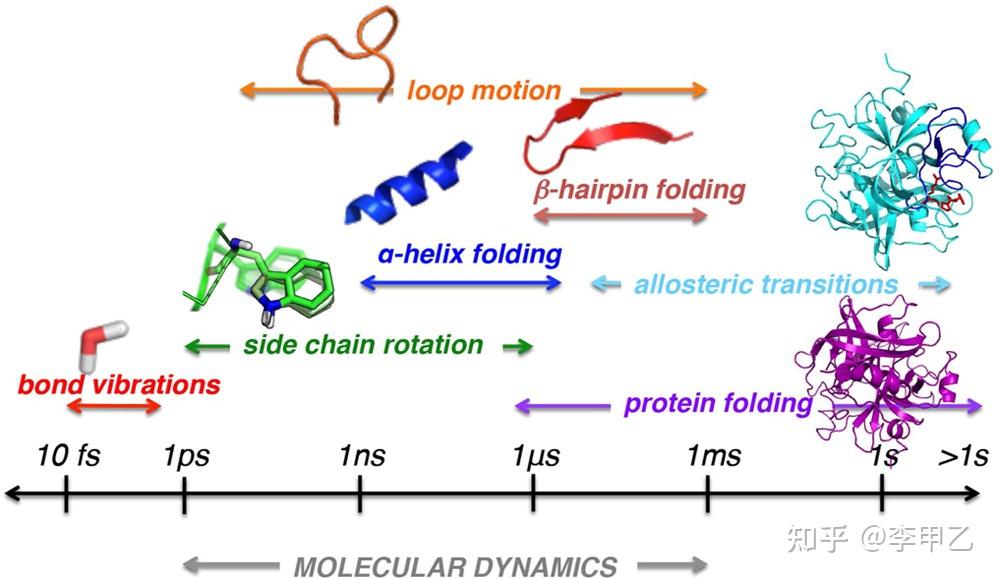
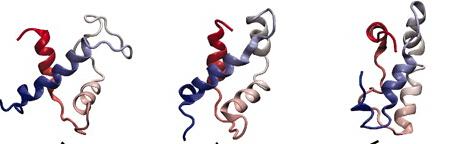
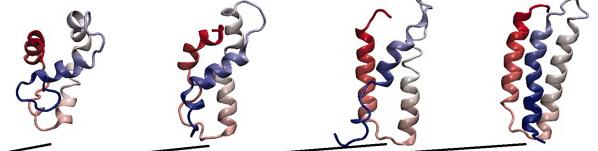
这一看就知道了,键振动(10–100 fs),侧链旋转(ps至μs),局部域波动(ns至ms),变构转变(μs至s)以及蛋白质结构motif的折叠( μs至s)。要注意蛋白折叠的时间尺度(us)和我们当今主流的算力跑几百ns是完全不一样的,那么我们怎么样有效地把模拟时间加长或者通过通过几百ns的模拟 去推测蛋白结构的变化呢,这是个难题1。
第二个很头疼的问题便是下图讲述的
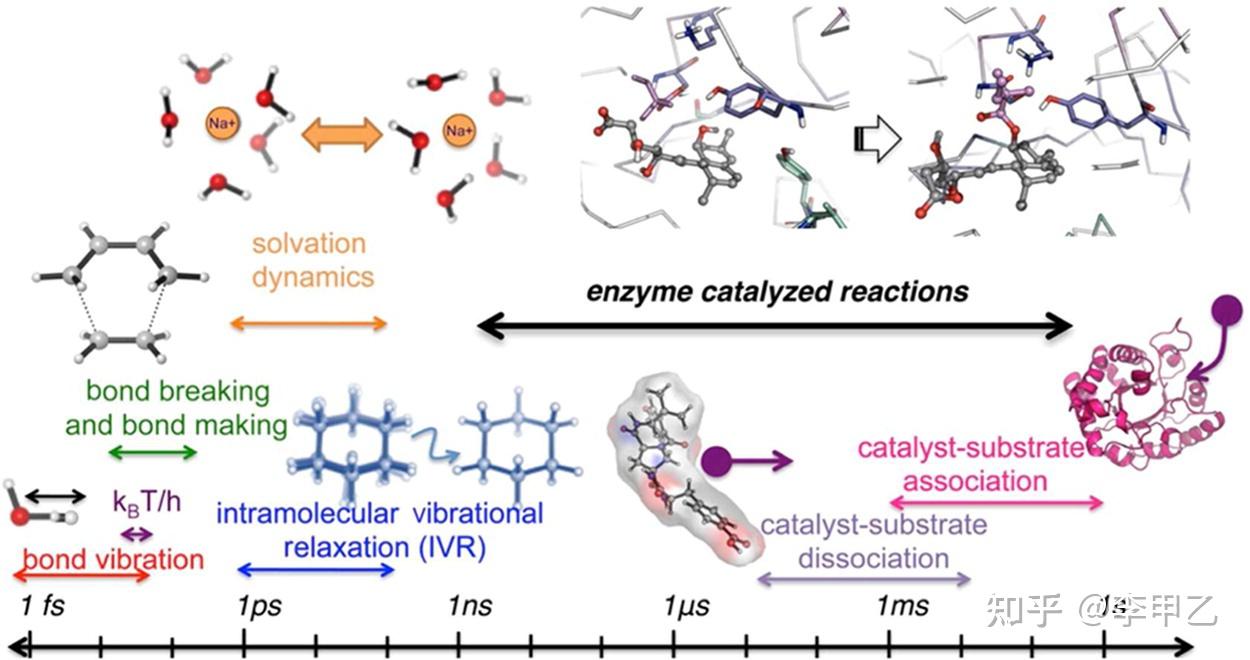
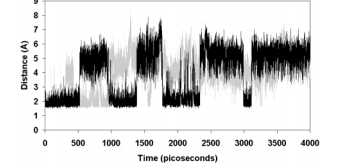
键断裂和形成的尺度(数十fs)比与蛋白质或溶剂碰撞或分子内振动弛豫的时标要短得多。所以一般的模拟,我们也无法去模拟键的断裂和形成,这是难题2。
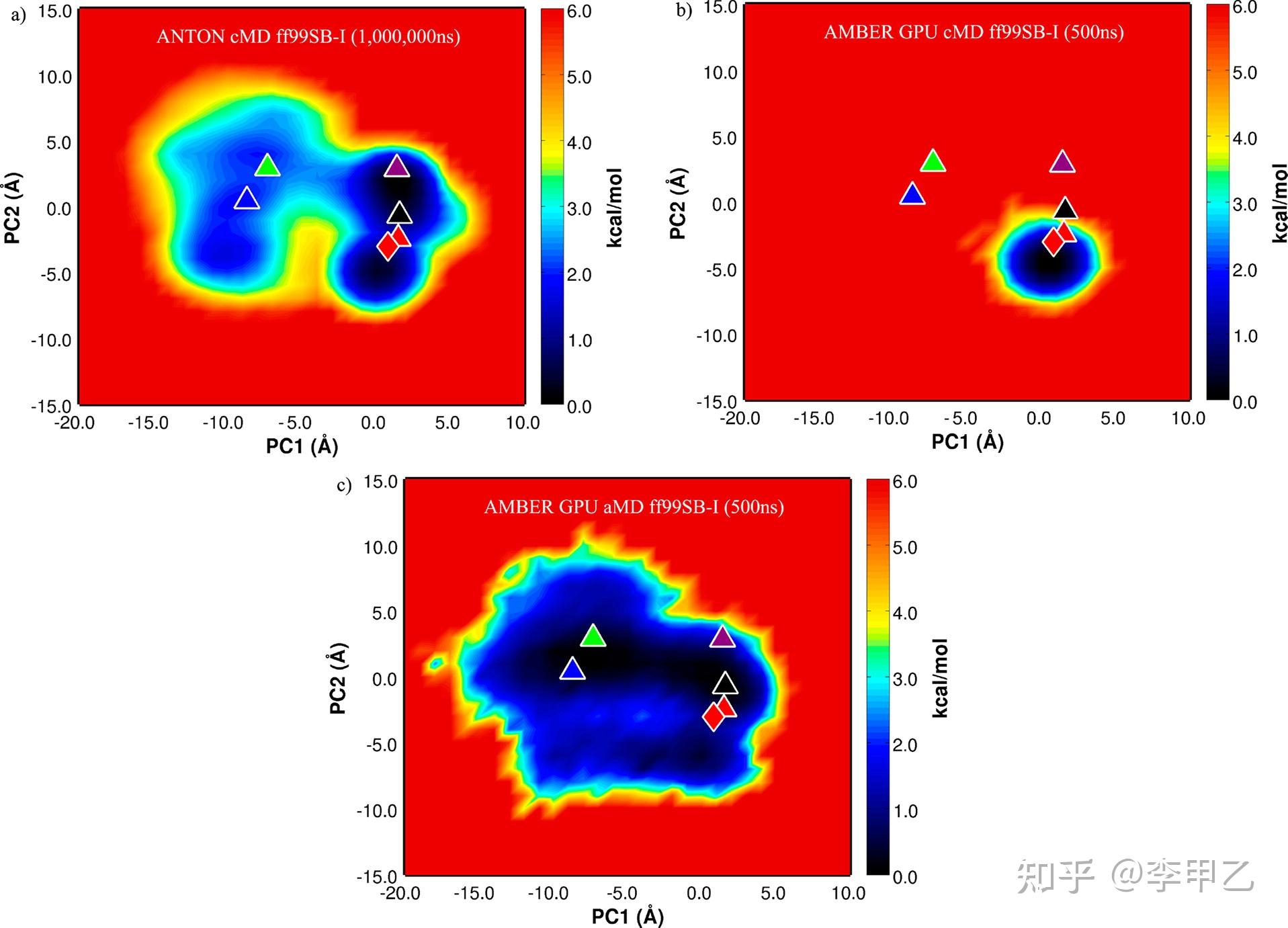
所以基于力场和基于 QM/MM 的 MD 模拟的空间采样应该足够使结果接近平衡分布。只有这样,才能应用统计力学机制,计算结合亲和力等特性,并与湿实验进行比较。最近十年,主要对于这两个难题,诞生了很多方法,以采样为例,包括靶向分子力学模拟(targeted MD, TMD、拉伸分子动力学模拟(steered MD, SMD) 加速MD(accelerated MD, AMD)、副本交换分子动力学模拟(replica exchange MD, REMD)以及 metadynamics, GaMD等等。我这里拿acceleratedMD(aMD)稍微展开下,aMD增强采样有2种方式,一是升高谷底,二是降低能垒,用500ns的尺度就可达到通常在毫秒级上发生的蛋白质构象变化,贴一个J. Chem. Theory Comput. 2012, 8, 9, 2997–3002的例子,达到这样的效果归功于结合GPU加速的能力以及aMD提高最小值的采样能力。
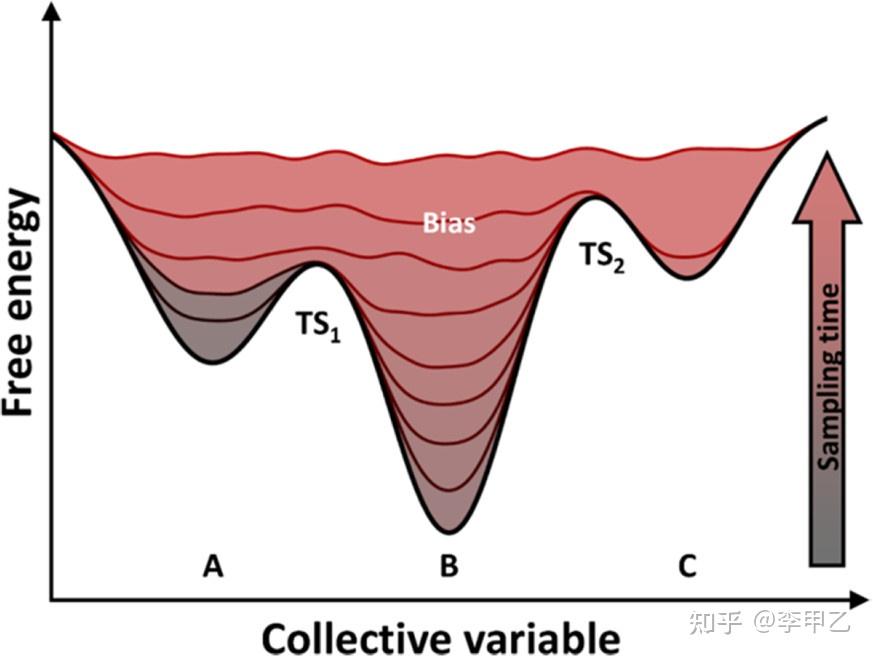
如果我们稳定住某些特定的感兴趣的构象的话,最近十年发展起来的有元动力学(metadynamics),SMD(steered MD),伞形抽样(umbrella sampling)等,这一块尤为重要,因为如果想知道自由能随特定反应坐标的分布情况. 这种分布被称为平均力势(PMF), 它对识别过渡态, 中间产物和终点的相对稳定性是非常有用的, 运行一次MD模拟来产生自由能随特定反应坐标的变化, 然后查看采样的概率, 但因为过渡态的能垒存在,比反应物以及产物都要高,所以MD模拟将仍然处在它一开始的局部最小值或者跨越不同的最小值, 但极少会采样到类似过渡态的结构. 采样的缺乏也就使我们不能产生一个精确的PMF,一种解决方法就是上面提到的伞形抽样umbrella sampling. 通过分解反应坐标成为一系列窗口, 然后采用限制迫使反应坐标维持在靠近窗口的中心。上述方法增强了 MD 的采样效率,但是它们仍然具有一定的局限,例如, TMD、 SMD 和 AMD 等方法通过引入外力而使初始结构在较短时间内实现向目标结构的转换,但外力的引入会使真实的转化路径发生偏移。
metadynamic yyds我觉得太厉害了!
另外AIMD必须有名字,基于量子化学方法的动力学一般称为从头算动力学(Ab initio molecular dynamics, AIMD),相比于基于一般的经典力场的动力学,其关键优势在于精度高、普适性强、能够描述化学反应,代价是耗时相差N个数量级。之前提到的ORCA有不错的做BOMD形式的从头算动力学的功能,使用很方便,而且本身ORCA做DFT的效率又高,是做孤立体系AIMD的首选程序之一。虽然有些特性不支持,比如没法像Gaussian的BOMD那样直接做准经典动力学,不能根据原子距离等标准判断什么时候自动结束任务等等,但都不是大问题。不过AIMD太烧机器了, 一般是作为QM之后对TDTS来NMA虚频统计采样后跑几百飞秒。CP2K 也能做AIMD,但CP2K的输入文件实在是太难写了。。(空了更)
这边说到TDTS 正好插播一条,也是近十年完善的理论基础,关于多步催化循环中能量模型,很长一段时间,其实计算化学和实验方之间缺乏一个有效的话语系统,计算化学往往聚焦在单步基元反应的能量势能面的变化,而生物学家则侧重表型如生物量、代谢流量、基因调控等。近十年来,Kozuch教授从相对简单的金属有机催化循环为研究对象阐明了多基元、多底物、多进程的催化循环的能量跨度模型,以蛋白质工程度整体催化循环效率的周转频率 TOF 为目标函数,而非单基元反应速率,从催化循环的全系统考虑各个参变量变化对蛋白质催化效率的影响。他利用Eyring 方程重构了基元反应速率 k 空间和势能面表示方法,提出了整合催化反应动力学与量子力学的催化循环模型,揭示了催化循环的周转效率 TOF 与反应能量校正后的能量跨度模型的关系,指出 TOF 的精确表达式与催化循环势能面中过渡态和中间的能量点贡献度,提出最高能量过渡态点和最高浓度中间体点和反应能来快速预测催化效率。从优化 TOF 的角度来讲,控制度接近 1的中间体和过渡态是决速中间体(TDI)和决速过渡态(TDTS)。个人觉得这理论如同物理数学定律是非常简洁而普适的的,相关文献我放两篇吧 文献的题目就很秀:第一篇How to Conceptualize Catalytic Cycles:The Energetic Span Model, Acc. Chem. Res. 2011, 44, 2, 101-110。第二篇:“Turning Over” Definitions in Catalytic Cycles, ACS Catal. 2012, 2, 12, 2787–2794
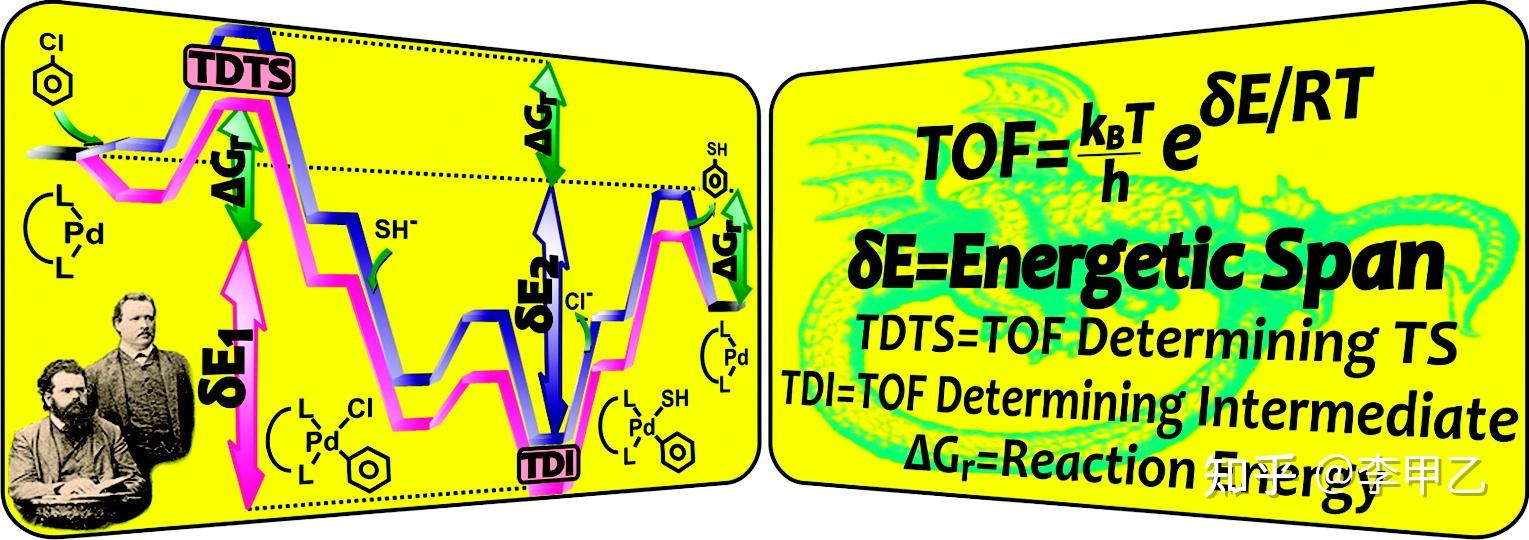
让我们继续说,很多时候,我们做MD模拟的目的就是想看看构象的变化,探究结构和催化行为的关系。
如果我们跑了很长时间的模拟,甚至可以构建马尔科夫模型(Markov state model简称MSM)最近几年很流行,文献奉上:Markov State Models: From an Art to a Science J. Am. Chem. Soc.2018, 140, 7, 2386-2396。MSM 通过构建跃迁几率矩阵的方式描述体系在不同亚稳态结构之间相互转换的概率,从而获得体系长时间尺度的热力学和动力学信息。MSM 是一个数学模型,它包含一个构象状态网络和一个描述这些构象之间转几率的转移概率矩阵 T。此模型的基本假设是时间 t 时的状态分布 X(t)足以确定经历时间∆t(∆t>0)后的状态分布 X(t+∆t),并且每个∆t 之后的状态分布 X(t+∆t)仅取决于当前的分布 X(t), 而与之前的任何状态无关, 这即为马尔科夫性也称为“无记忆”性。构建MSM模型,一般要选择n个状态,使得它们涵盖了整个动力学行为,并且滞后时间τ足够长以成为马尔可夫模型,但又短得足以解决系统动力学问题。下面是一般的流程:
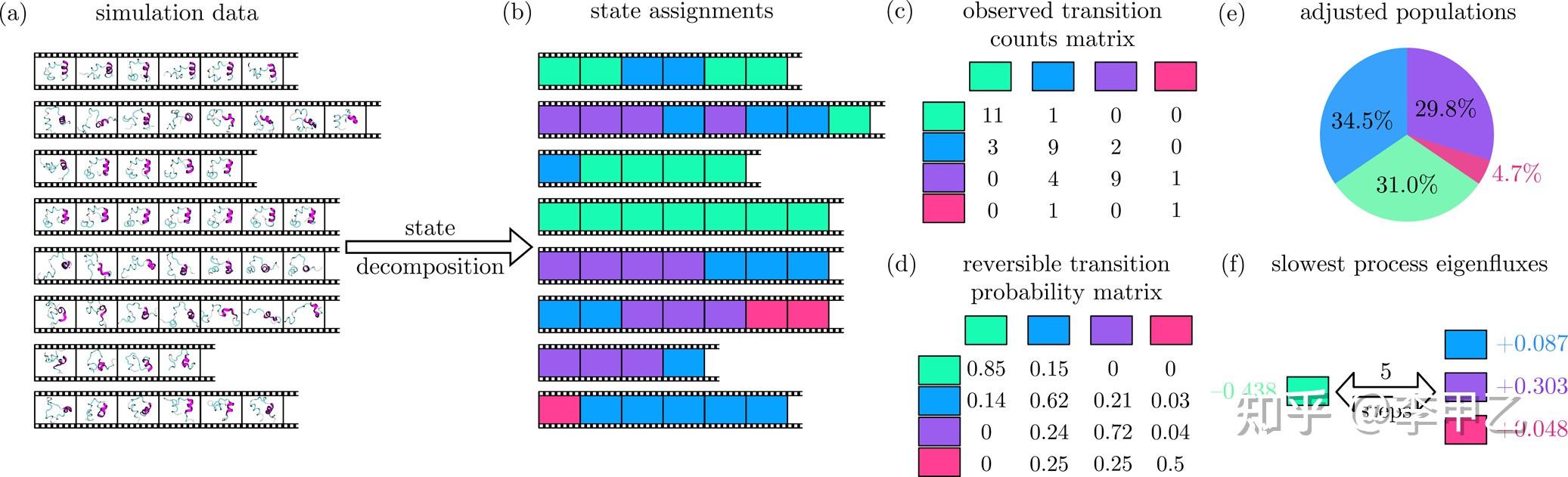
我觉得比较不错的方法就是通过细分再聚类的方法构建 MSM。首先对 MD 轨迹进行降维处理,所用方法为时间-结构独立成分分析(time-structure independent component analysis, tICA)。然后再将降维后的数据聚类成数百个微态,所用方法为 K-Centers;最后,再根据力学性质将微态进一步划分为若干亚稳态,所用方法为 PCCA(Perron cluster cluster analysis)。
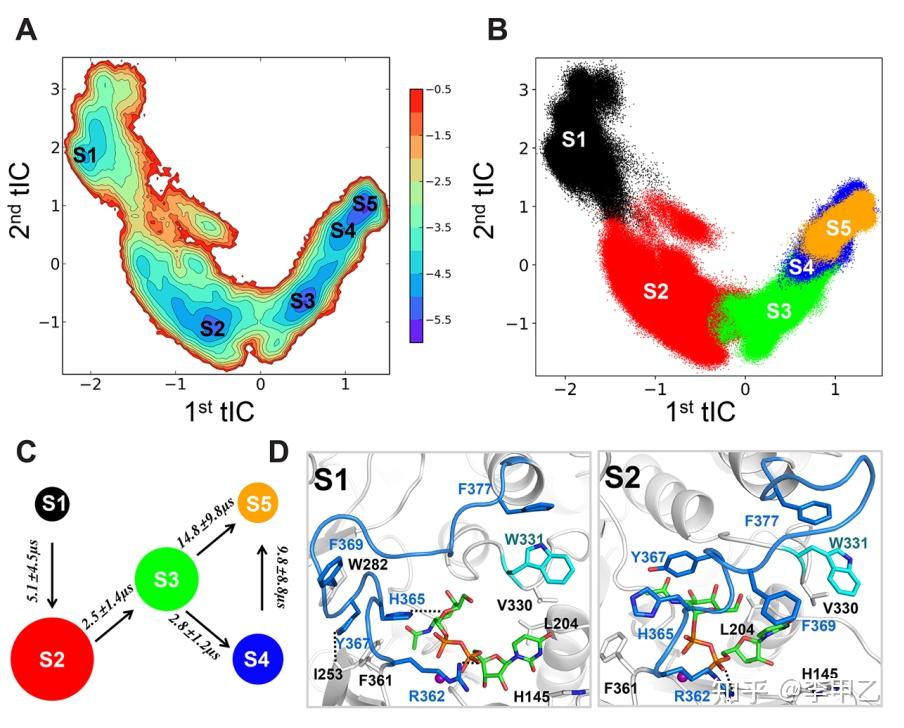
往大了说 我们可以利用MD模拟得到轨迹数据进行无监督学习
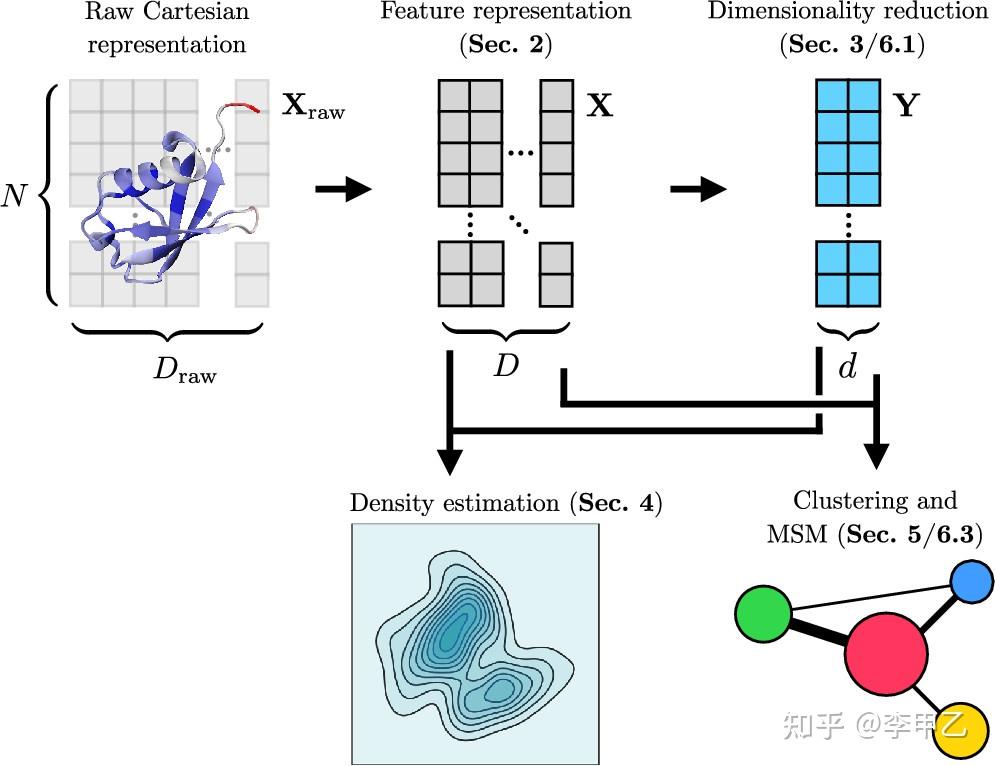
3.3 结合自由能的计算
我决定专门在这个版本讲讲 近十年结合自由能计算的发展和突破
我经常遇到药物化学或是生物化学相关的实验组来询问我,蛋白-配体相互作用,蛋白-蛋白相互作用的问题,确实,大多数药物通过作用于特定的生物靶标发挥作用。能否准确预测药物和靶标之间的相互作用,是实现分子设计的一个关键问题。在文献Journal of Medicinal Chemistry, 2016, 59, 4033中就提到准确计算药物-靶标的结合能是该领域中过去30年以来的圣杯,现在也仍是这样。
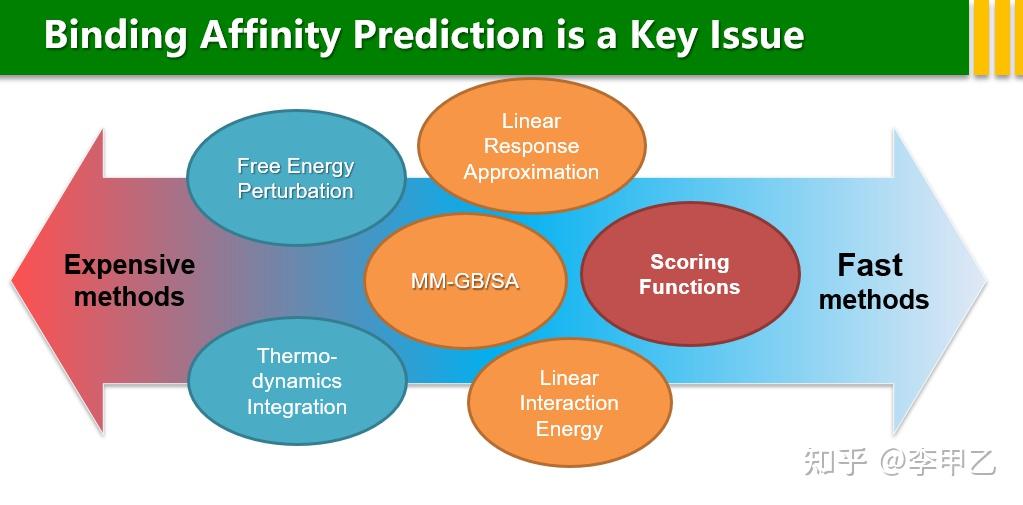
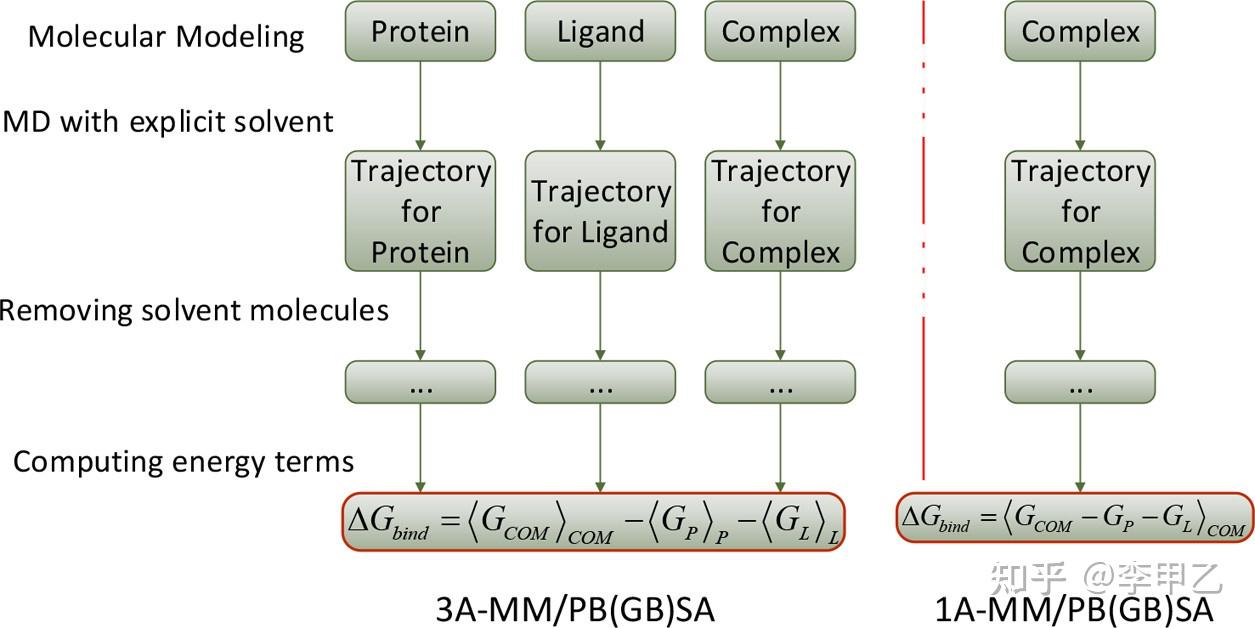
上世纪80年代以来,出现了各种门类的理论方法用于定量计算蛋白-配体亲合性,很多商业的软件比如薛定谔,discovery studio,MOE等, 以及最近几年发展的数十种各种对接软件,Autodock,HADDOCK, ZDOCK,RosettaDock server都提供了打分函数,还有很多网页版的,方便做实验的同学快速计算,比大小 就知道结合的趋势。打分函数一般作为结构的初筛,然后再用Amber或者Gromacs跑MD,用MM/GB(PB)SA的方法计算结合自由能,这种方法的误差也有10kcal/mol,还是蛮大的,实际应用中常见的形式的是  Gbind,因为这是最常见的,我贴一个计算流程,国内做的不错的比如来自浙大的侯廷军组,侯老师回国后从2011年开始致力于相关的研究,发表了一系列Assessing the Performance of the MM/PBSA and MM/GBSA Methods的文章,在2020年底时,J. Chem. Inf. Model期刊发表了一个专刊,其中收录了10篇 60年来在JCIM发表的最具影响力的研究论文,其实就有侯老师的一篇:J. Chem. Inf. Model. 2011, 51, 1, 69-82 Gbind,因为这是最常见的,我贴一个计算流程,国内做的不错的比如来自浙大的侯廷军组,侯老师回国后从2011年开始致力于相关的研究,发表了一系列Assessing the Performance of the MM/PBSA and MM/GBSA Methods的文章,在2020年底时,J. Chem. Inf. Model期刊发表了一个专刊,其中收录了10篇 60年来在JCIM发表的最具影响力的研究论文,其实就有侯老师的一篇:J. Chem. Inf. Model. 2011, 51, 1, 69-82

我们知道分子对接的预测能力依赖于其所用蛋白−配体打分方法的预测精度,MM/PB(GB)SA方法已成为近10年应用最为广泛的结合自由能预测方法之一,但其在虚拟筛选中的总体预测能力和应用范围一直未有定论,阻碍了其在创新药物发现中的应用。于是对6个蛋白质体系的59个配体小分子的结合自由能进行了系统的评估研究,研究了分子动力学模拟时间、溶质介电常数、构象熵的计算以及不同PB和GB模型对预测结果的影响。研究确定了介电常数和靶点–小分子界面性质之间的定量关系,为自由能计算中介电常数的选取提供了比较可靠的依据。研究还发现,MM/PBSA的预测结果更接近实验数值,而MM/GBSA则对相对结合自由能预测性能更优。在后续的研究中,还深入探讨和评估了分子力场、溶剂化模型、原子电荷、构象熵、动力学采样时间等多种因素对MM/GBSA预测精度的影响。这里贴一篇前年发表的综述,对MM/GB(PB)SA方法感兴趣的可阅读:Chem. Rev. 2019, 119, 16, 9478-9508。
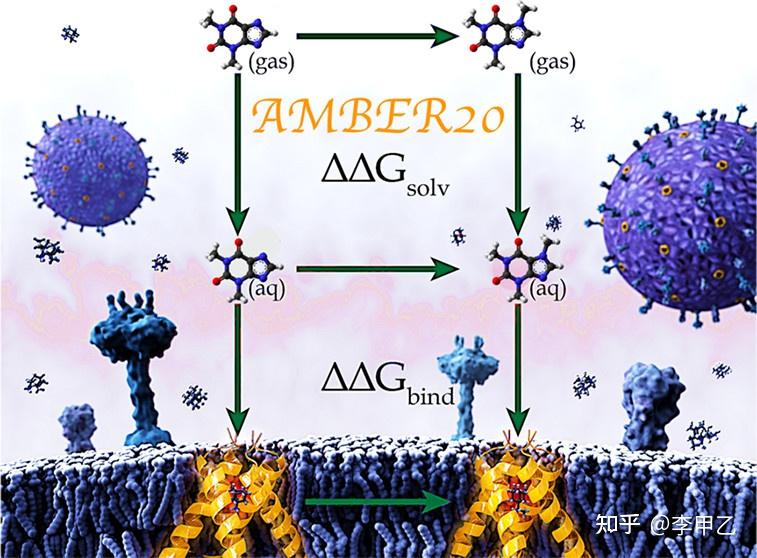
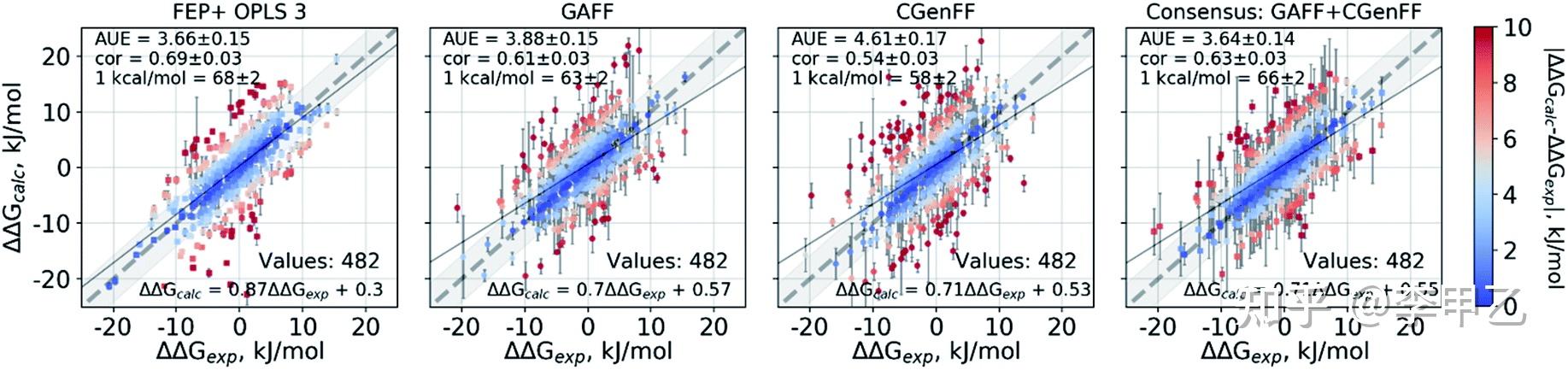
然后如果精度再往上走,误差达到1~2kcal/mol的计算方法,就要涉及炼金术(Alchemical)结合自由能的计算了,热力学积分(Thermodynamic Integration ),自由能微扰(free energy perturbation)等就属于这个范畴,很多用荧光偏振实验,IC50实验测定差异不大的,在计算上重现就要上用方法,药化,生化相关专业的同学值得关注。炼金术自由能计算涉及热力学循环的概念,从上世纪80,90年代开始有人做,到近十年通过Gpu加速计算,使得这种昂贵的精度极高的方法越来越普及,对于小分子官能团的一系列改变,以及蛋白残基的一系列突变都能进行。由于自由能是一种状态函数,热力学终态(结合和未结合)可以通过任何途径连接起来,在实践中可以构建多条热力学循环路径,利用可以优化计算的最终状态之间的“炼金术”途径。Gromacs和Amber都可以跑,用商业的薛定谔软件FEP+也可以。我用的最多的就是Amber,大概从AMBER16开始,应用Alchemical方法的文献是越来越多, 在2014,2015,2016年,用此技术方法纯计算发JACS还是可以的。在计算中容易遇到的问题有三种:“终点灾难”、“粒子崩溃问题”和“大梯度跳跃问题”,比如终点灾难就比较常见,在热力学终点(λ 值接近 0 和 1),dV/dλ的数值会发生跳跃。
3.4 自由能计算
先空着
QM/MM的发展
我们知道分子力学(MM)精度低,不适合描述化学反应,但可以处理很大的体系,QM精度高,但能处理的体系较小,将两者结合,就可以将体系最重要的区域用QM,不重要的区域用MM,就可以处理尺度介于QM和MM之间的体系,比如溶质和溶剂的体系,酶催化的反应等等。
这个思想是1976年就有了,就是2013获得诺奖的三个大佬弄的

The Nobel Prize in Chemistry 2013 was awarded jointly to Martin Karplus, Michael Levitt and Arieh Warshel "for the development of multiscale models for complex chemical systems.
近十年,由于计算能力的提升,使得QM/MM方法成为十分广泛的手段,实现QM/MM的程序也很多,比如Gaussian就支持AMBER,UF力场,以ONIOM方式将QM与MM结合做优化,振动分析等;Q-chem也能做,支持AMBER,OPLS-AA,CHARMM力场;ORCA能做,支持把CHARMM力场转化成自身格式然后做QM/MM单点,优化,找TS,IRC,动力学等。CP2K可直接做QM/MM的优化和动力学,然后也有如Chemshell这样的接口程序,将分子动力学程序GROMOS,GULP,CHARMM与量化程序Gaussian,ORCA,Dmol3,Q-Chem相连接做优化,动力学等。对于酶催化的很多机理探究,协同反应,分布反应,从反应物 到产物释放,我们往往就是先跑MD,跑完MD模拟之后,计算 QM/MM反应势垒来确定多种反应途径之间的竞争关系,最常用的是ONIOM方法,以及最近几年发展起来的簇模型(Cluster model)。
想了解ONIOM的 可以看Chem. Rev.2015, 115, 12, 5678–5796:The ONIOM Method and Its Applications
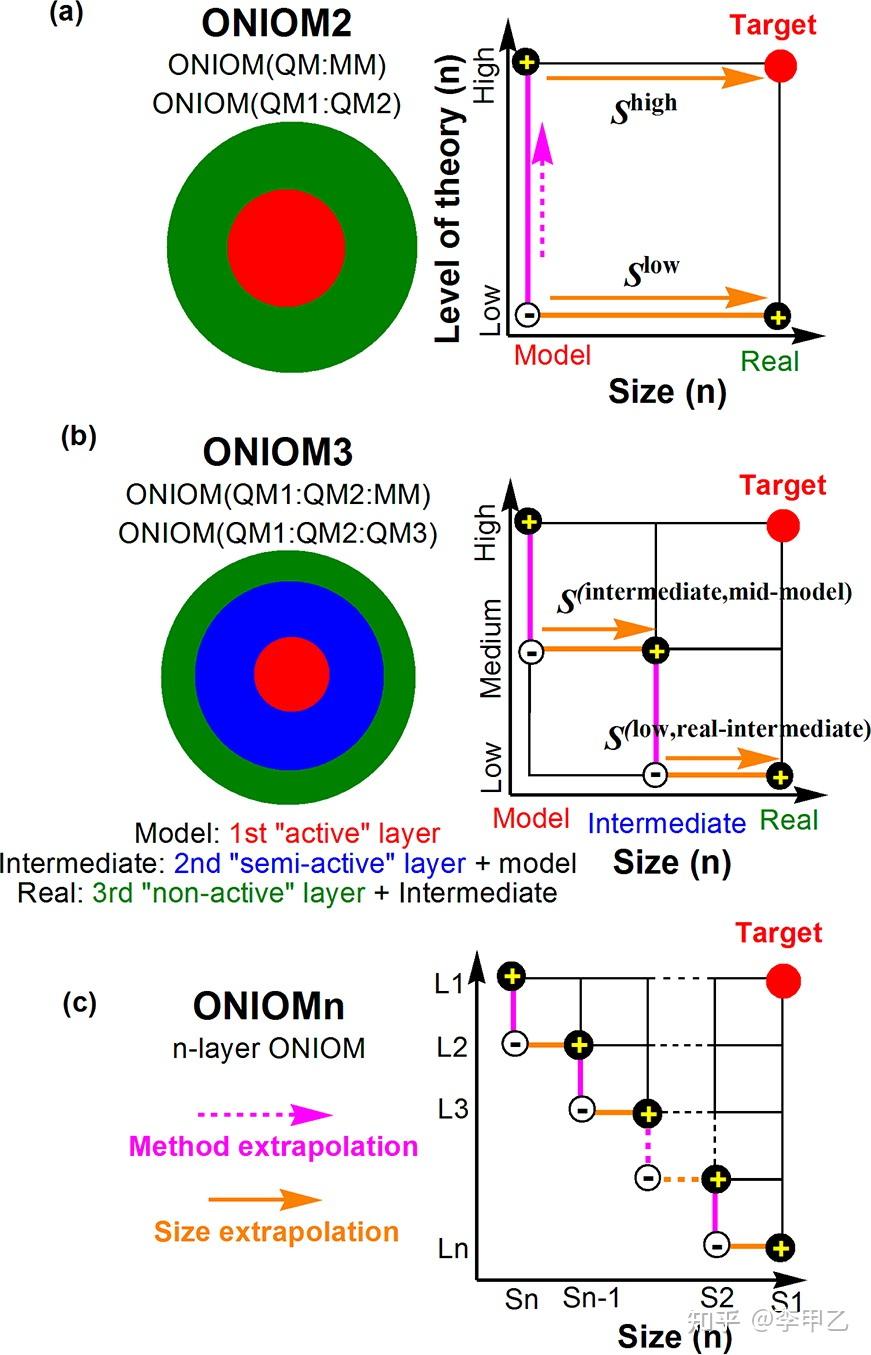
个人用起来 觉得比较麻烦的就是1:边界原子的处理,2:补全参数,也像TAO包,这样的Gaussian做ONIOM的辅助工具,包含了一套Perl脚本文件来构建输入文件,监控计算,还是不错的。
想了解簇模型的 可以看Fahmi Himo组的文章,他们组不用ONIOM方法,一律用簇模型,因为用ONIOM(QM:MM)方法计算酶催化等生物体系时,通常的处理会讲反应位点取做QM区域,周围一定距离的残基取作MM区域,而簇模型也叫all-QM或者QM-only模型,选取的原子数会多一些,大概200-300个。列举两篇:Recent Trends in Quantum Chemical Modeling of Enzymatic Reactions. J. Am. Chem. Soc. 2017, 139, 6780-6786以及Modeling Enzymatic Enantioselectivity using Quantum Chemical Methodology ACS Catal. 2020, 10, 11, 6430–6449.
4.突破性的发展
我发现有些进展和方法 既不属于QM,或者MD,QM/MM,于是我设置了这一章节,从何讲起呢?就从2018年的诺贝尔化学奖开始吧,我直接引用官网的文字:The Nobel Prize in Chemistry 2018 was divided, one half awarded to Frances H. Arnold "for the directed evolution of enzymes", the other half jointly to George P. Smith and Sir Gregory P. Winter "for the phage display of peptides and antibodies."

阿诺德的领域就是涉及酶的定向进化(Directed Evolution)。最初,引入了随机氨基酸变化的迭代循环,随后选择了具有改进的热稳定性,底物特异性和对映选择性的变体。不仅在实验上批量突变筛选选择,计算化学上也因为分子动力学方法和技术的飞速发展,使得对酶的研究高速推进,我列举一些高速发展的分支:酶的机制和选择性,新型催化酶的预测,立体选择性有机反应,催化剂的机理和设计,有机金属反应机理等等,每个分支还有很多二级三级的分支,我很难对每一块都像写综述一样去介绍和列举,就挑一些典型作为例子,抛砖引玉。
4.1 理论酶(theozyme),
如果你从事有机化学或者对有机化学有些了解,便知道取代反应,自由基反应,加成反应,消除反应,氧化还原反应,各种重排反应,看得直接头大,但在生物体内的化学反应,种类其实并不多,又因为酶的高效性,计算上就能通过计算模型官能团去稳定过渡态的最佳几何构形 去构建,像造飞机造汽车一样去设计改造enzyme. 这一块的大佬我列举三位:K. N. Houk;Donald Hilvert;David Baker
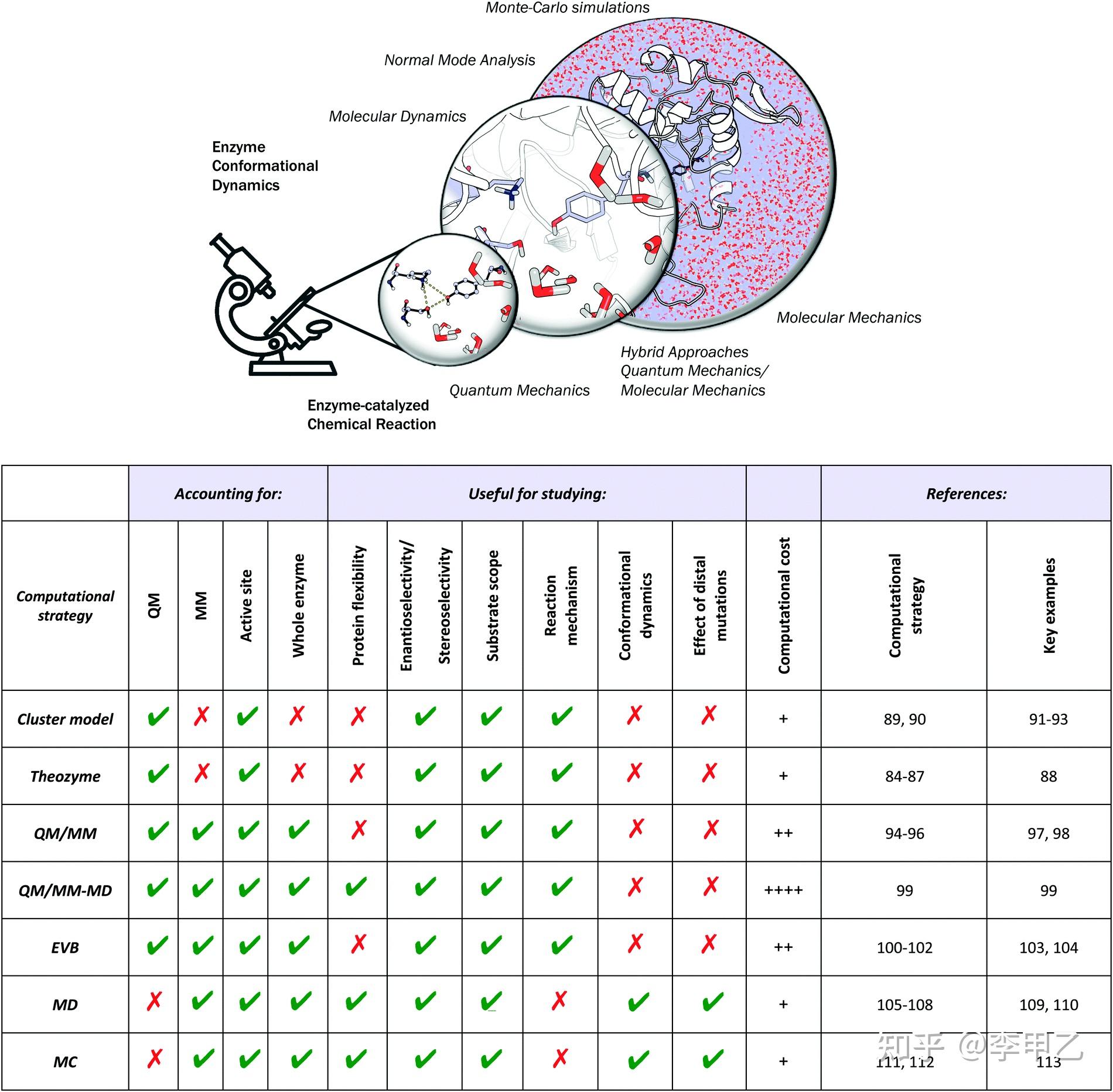
顺着定向进化说,如何通过氨基酸残基突变优化在新底物和新环境下高活化能反应势能面的生物合成酶?实验科学家通过多轮定向进化对氨基酸序列进行随机性的优化和改进。但是计算化学领域的科学家则提出了诸多催化的模型 用于生物合成酶和工业酶的理性设计和优化。通过对反应势能面上反应物、产物、过渡态、中间体等驻点(能量极值点)的计算来透彻刻画酶催化反应机理,理论学家基本认同了酶具有将反应的总活化能(或表观活化能)减低到 15-20 kcal/mol 的能力,等价于半反应时间缩短到毫秒 (millisecond) 至秒 (second) 的范围。
对蛋白质分子如何降低总反应活化能的机制理解上存在一些不同的看法,比如上面提到的来自UCLA的Houk教授就认为,类似有机化学催化反应,酶催化能力主要来自蛋白质的特殊氨基酸残基稳定了催化过程中的关键过渡态具体的计算一般是了解酶活中心催化的机理,一个生化反应有非常多的可能途径,特定蛋白质分子的结构具有降低关键过渡态的作用,可以通过计算底物与周边关键氨基酸残基复合体 Theozyme模型来理解,然后利用所获得的过渡态与周边残基复合物的几何结构利用inside-out 等策略构建蛋白质框架,来开发和优化新酶。我就举两个例子,第一个是水解酶,很多会有个Ser-His-Asp的催化三联体,或者Cys-His这样的催化二联体,以及一个氧离子穴,Ser或者Cys就可以作为亲核试剂,并通过氢键 His去质子化,就能进攻底物的酯键的C原子,形成一个四面体,His上的H会和底物的C=O的O原子结合 释放醇,这就完成了酰化的步骤,接着就是水分子的进攻 水解,这样一轮反应cycle就完成了。知道机制后 就可以去设计改造酶了
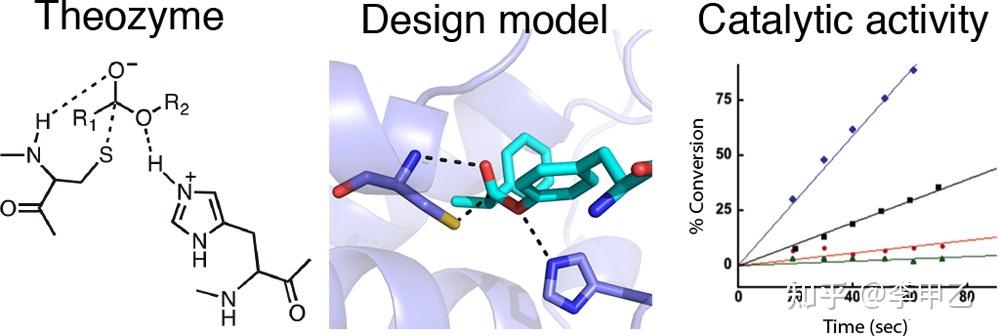
J. Am. Chem. Soc. 2012, 134, 39, 16197–16206 ; Nat Chem Biol. 2014;10(5):386-391.
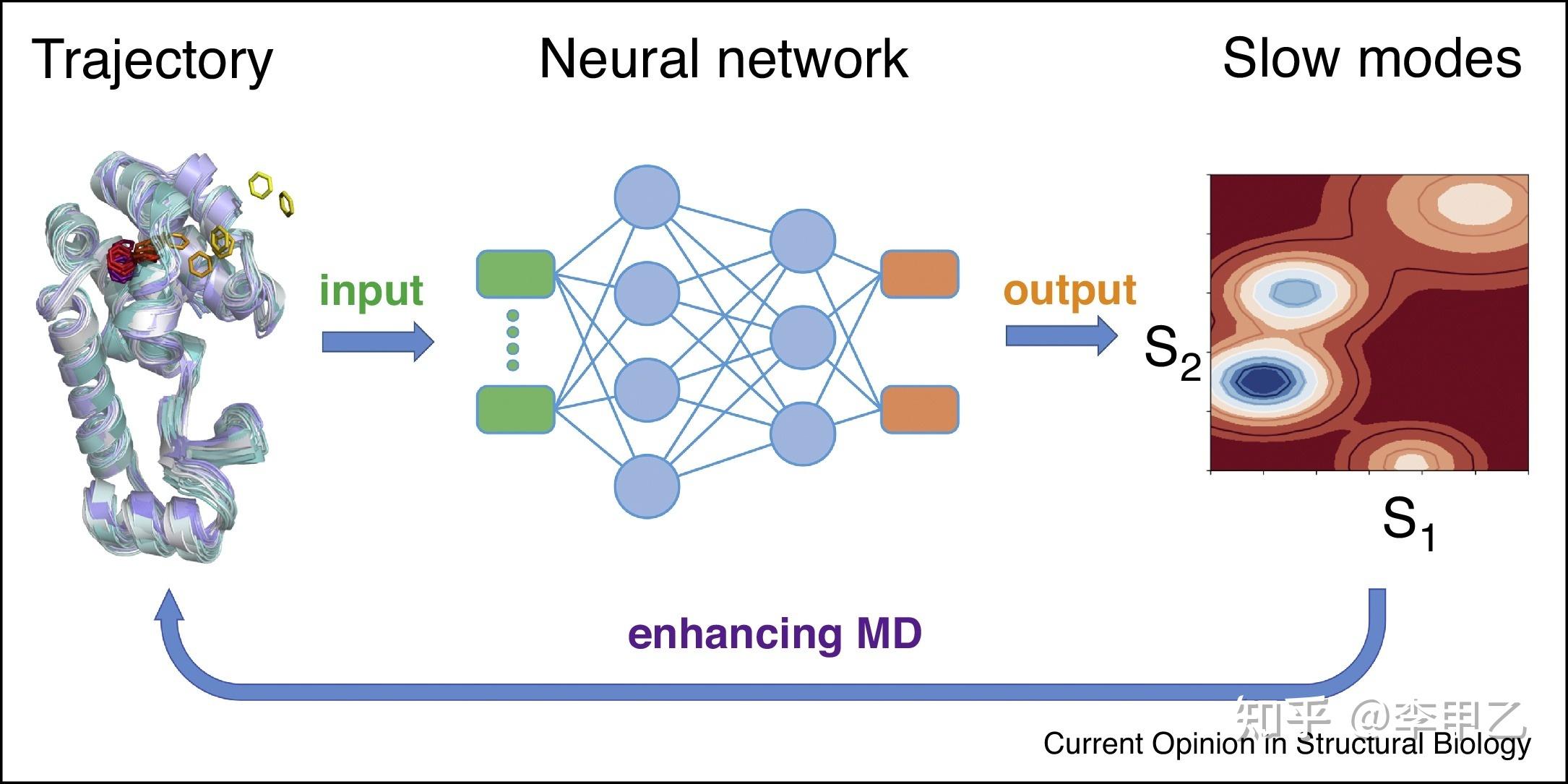
4.2 机器学习
很多时候在MD模拟中的ML,指结合人工神经网络(ANN)的方法,这方面先起个头
5.波函数分析软件Multiwfn
Multiwfn是极为流行、功能最全面的的量子化学波函数分析程序。通过现成的量子化学程序,在各种近似条件下求解薛定谔方程,就可以得到体系的波函数。波函数包含了体系中一切信息,可以视为是个黑箱,通过对波函数及其衍生信息(如电子密度)进行各种方式的分析,就可以从这个黑箱中提取各种具有化学意义的信息,使体系的内在特征能够被我们透彻地认知,诸如原子间键的强度与本质、电子在体系中的行为和分布方式等,同时也可以预测当前体系如何与其它体系相互作用,诸如预测反应位点。
因为十分容易上手,十分强大,被引率快破万了,在此感谢sobereva大大
 |
|

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 






 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 发表于 2025-3-10 13:38
发表于 2025-3-10 13:38