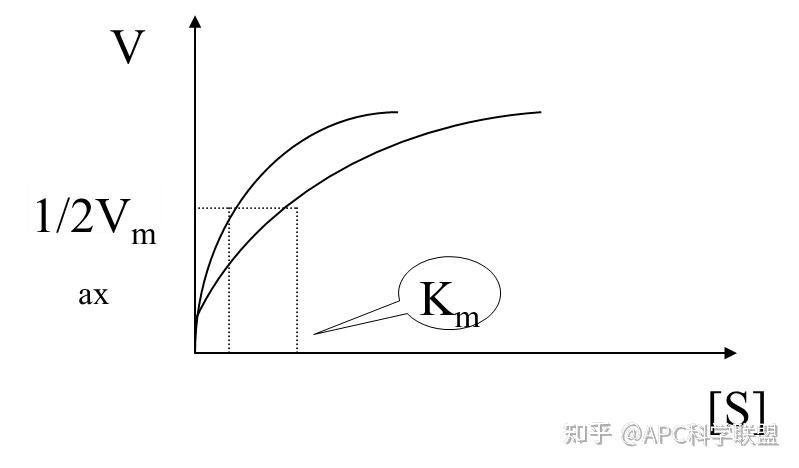
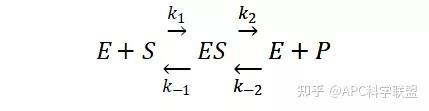
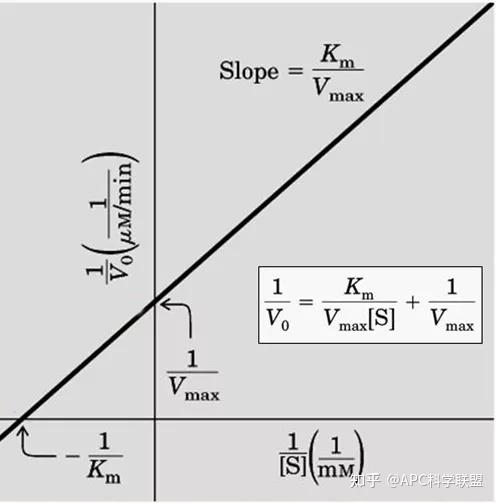
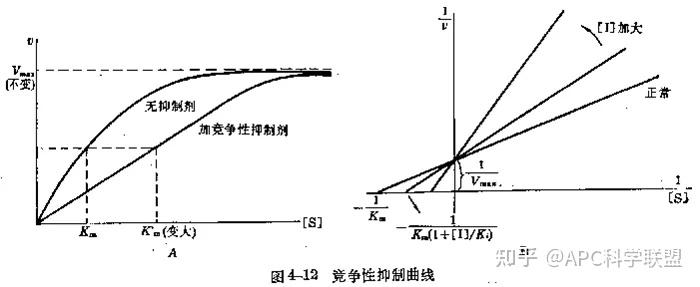
其中V0为酶促反应的初速度,Vmax为酶促反应的最大速度,[S]为底物浓度,而Ks为解离常数,即K2/K1 米氏方程的推导建立在三个基本假设上:
(1)在反应开始初期,产物的生成量极少,逆反应可不予考虑;
(2)底物浓度远远大于游离酶的浓度,因此在反应过程中底物浓度基本上保持恒定;
(3)反应的第二步为反应的限速步骤,即K2 ≫ K3,以保证 ES 分解为 P 的速率不足以破坏 E 和 ES 之间的平衡。
其推导过程如下:
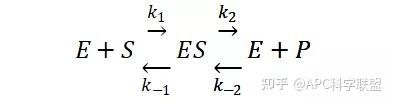
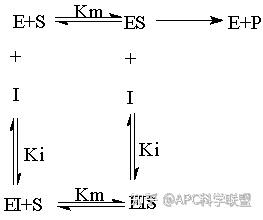
假定 E + S → ES 可迅速建立平衡,且反应的第二步相比第一步要慢得多,因此,该反应的整体反应速率由第二步决定,即:
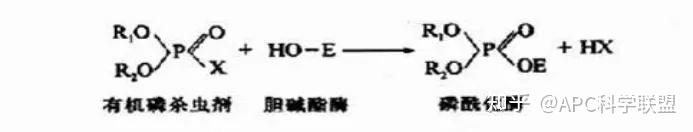
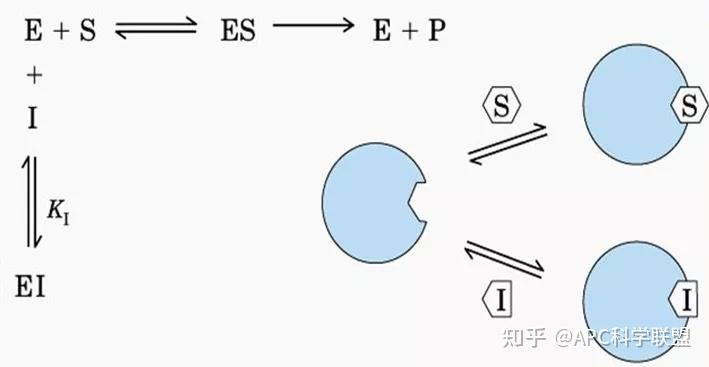
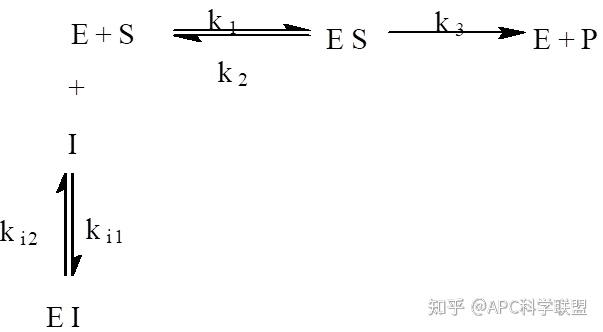
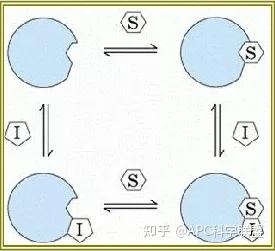
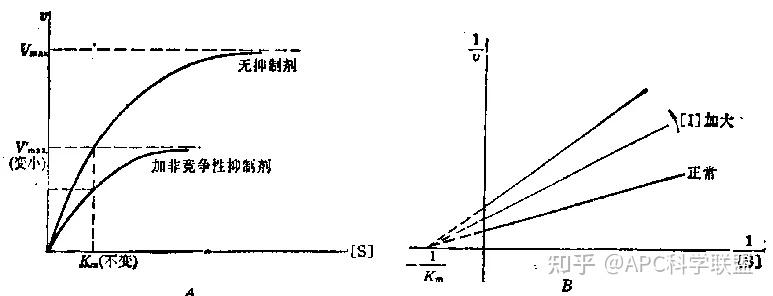
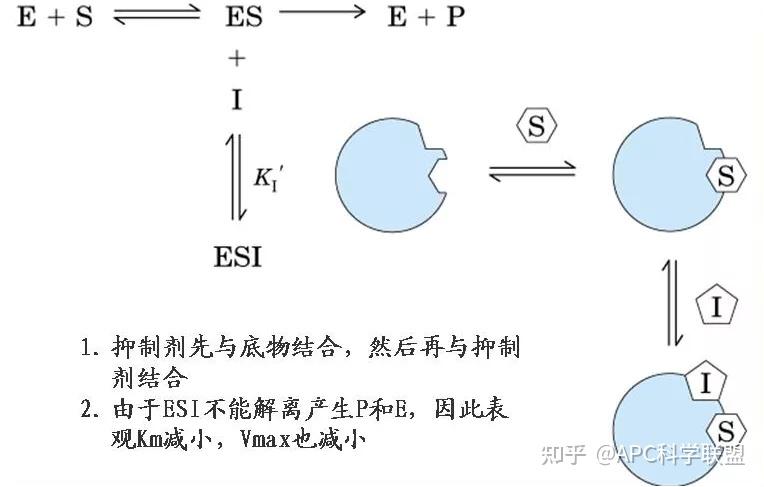
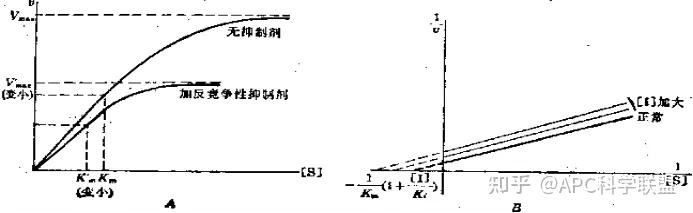
非竞争性抑制剂可与酶活性中心以外的基团结合,引起酶分子的构象变化使之不能催化底物形成产物。非竞争性的抑制剂既可以与游离酶结合,又可以与底物-酶复合物结合形成 EIS 三元复合物。抑制剂的结合不影响酶与底物的结合,但会阻止酶-底物复合物进一步反应生成产物,因此酶活性降低。一些重金属离子如Cu2+、Pb2+、Hg2+的抑制作用即属于此类

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡 发表于 2025-3-11 15:41
发表于 2025-3-11 15:41


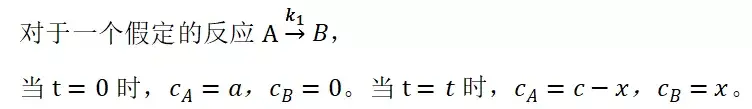
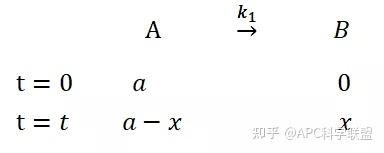
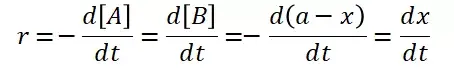
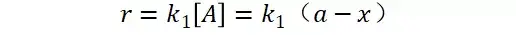
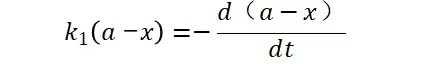

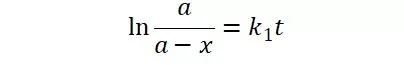

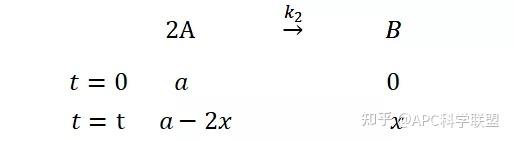
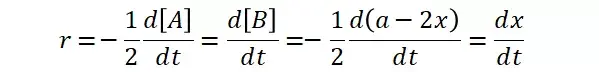
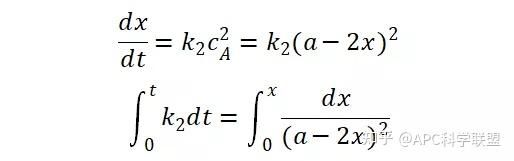
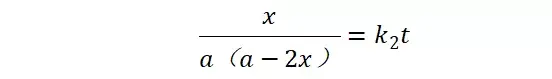
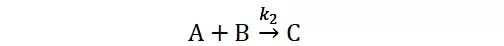
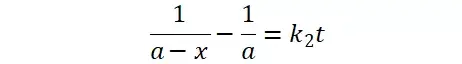
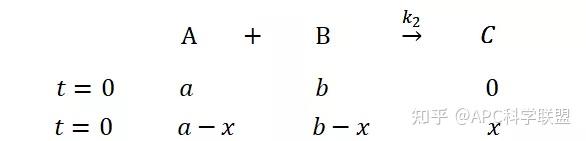

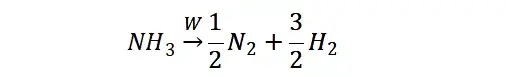


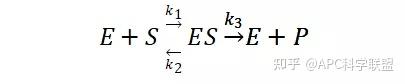

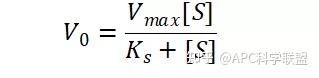
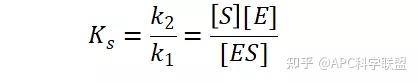
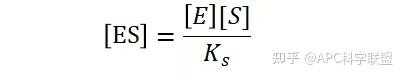
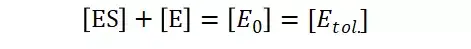
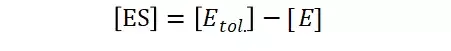
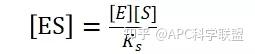
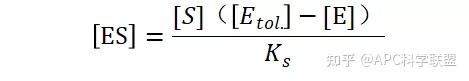
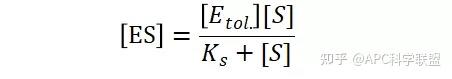
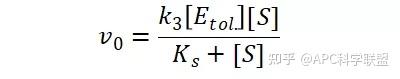
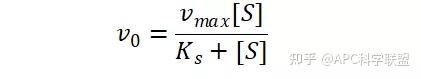
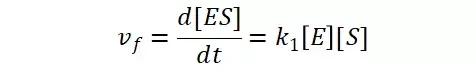
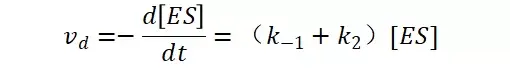
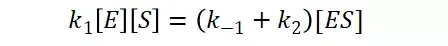
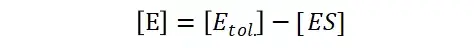
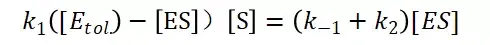
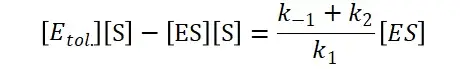
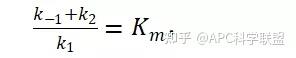
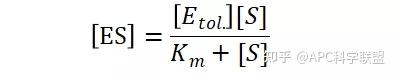
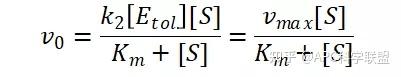
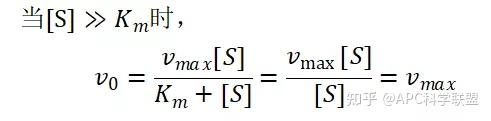
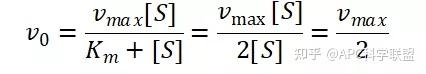
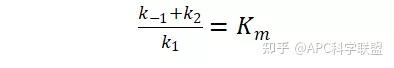

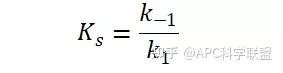
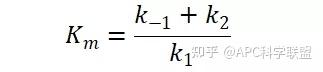
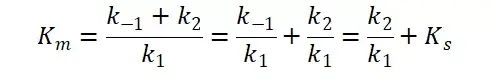
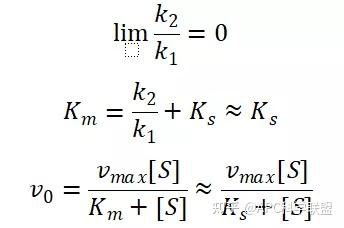
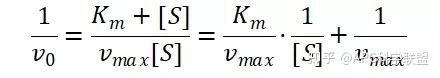
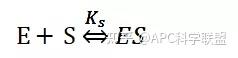
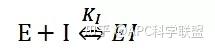
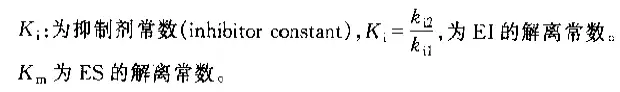
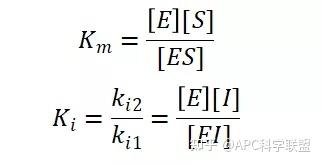

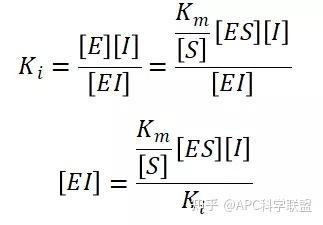
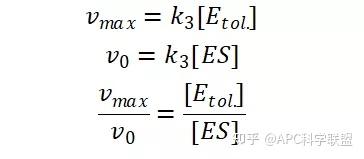
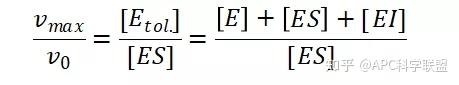
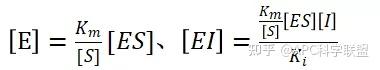
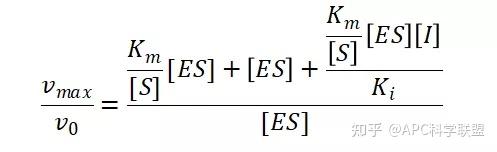
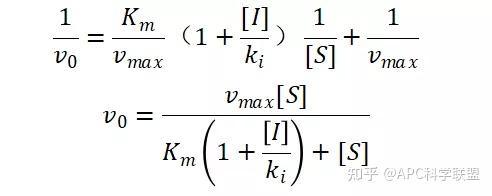
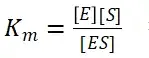
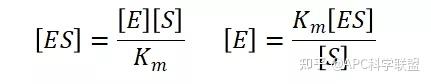
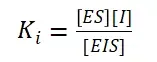
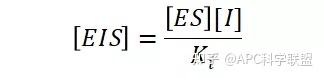
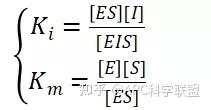
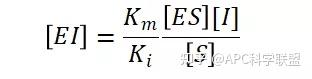
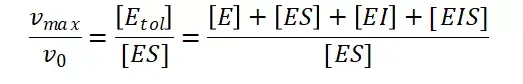
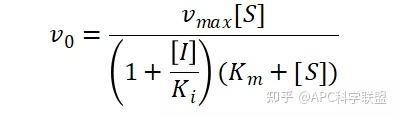
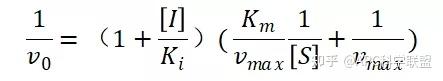

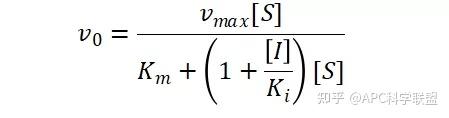
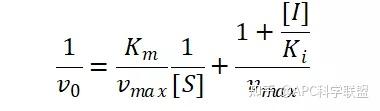
 提升卡
提升卡


