用户名
UID
Email
密码
记住
立即注册
找回密码
只需一步,快速开始
微信扫一扫,快速登录
开启辅助访问
收藏本站
快捷导航
门户
Portal
社区
BBS
资讯
会议
市场
产品
问答
数据
专题
帮助
签到
每日签到
企业联盟
人才基地
独立实验室
产业园区
投资机构
检验科
招标动态
供给发布
同行交流
悬赏任务
共享资源
VIP资源
百科词条
互动话题
导读
动态
广播
淘贴
法规政策
市场营销
创业投资
会议信息
企业新闻
新品介绍
体系交流
注册交流
临床交流
同行交流
技术杂谈
检验杂谈
今日桔说
共享资源
VIP专区
企业联盟
投资机构
产业园区
业务合作
投稿通道
升级会员
联系我们
搜索
搜索
本版
文章
帖子
用户
小桔灯网
»
社区
›
C、IVD技术区
›
ELISA技术
›
IF=14.6,学术圈的绝配CP“泛素化+自噬,发文好思路 ...
图文播报
2026庆【网站十三周
2025庆【网站十二周
2024庆中秋、迎国庆
2024庆【网站十一周
2023庆【网站十周年
2022庆【网站九周年
返回列表
查看:
6595
|
回复:
0
[分享]
IF=14.6,学术圈的绝配CP“泛素化+自噬,发文好思路
[复制链接]
007
007
当前离线
金桔
金币
威望
贡献
回帖
0
精华
在线时间
小时
雷达卡
发表于 2025-3-2 07:30
|
显示全部楼层
|
阅读模式
登陆有奖并可浏览互动!
您需要
登录
才可以下载或查看,没有账号?
立即注册
×
今天探长给大家分享的文章非常新颖!听探长给大家介绍一下这篇文章的魅力所在吧!
1、研究内容新颖,首次发现MLKL与自噬调控之间的联系,并揭示了其分子机制。这一发现拓展了MLKL的功能,不仅在坏死过程中发挥作用,也在维持细胞正常生理功能方面起关键调控作用。
2、研究系统全面,结合了分子生物学、生化、行为学和病理学多方面的证据,论证过程严谨,结果可靠且具有说服力。
3、研究利用多种方法技术手段,如CRISPR/Cas9敲基因小鼠模型、单个细胞RNA测序、质谱分析等,研究方式先进。
4、研究结果具有重要的病理意义,提示可以通过调控MLKL来达到调节自噬、维持蛋白质稳态的治疗目的,为防治神经退行性疾病提供了新视角。PS:这篇文章思路清晰内容完整,如果你也有想法但不知道从何下手,那就抓紧联系探长!
https://u.wechat.com/ENOSAvW697E5iWQsr4zvEaI (二维码自动识别)
题目:MLKL-USP7-UBA52信号通路通过维持泛素稳态,是大脑自噬过程中不可或缺的信号通路
杂志:Autophagy
影响因子:IF=14.6
发表时间:2024年9月
研究背景
假激酶(MLKL)是细胞坏死中的关键效应分子。个体存在MLKL基因缺失会增加患神经退行性疾病如阿尔茨海默病的风险。但是MLKL与神经退行性疾病的关系机制尚不清楚,需要进一步研究。本文通过在小鼠和细胞模型中敲除MLKL基因,发现缺失MLKL会抑制自噬过程。
研究思路
研究结果
1.自噬在没有假激酶的情况下被抑制
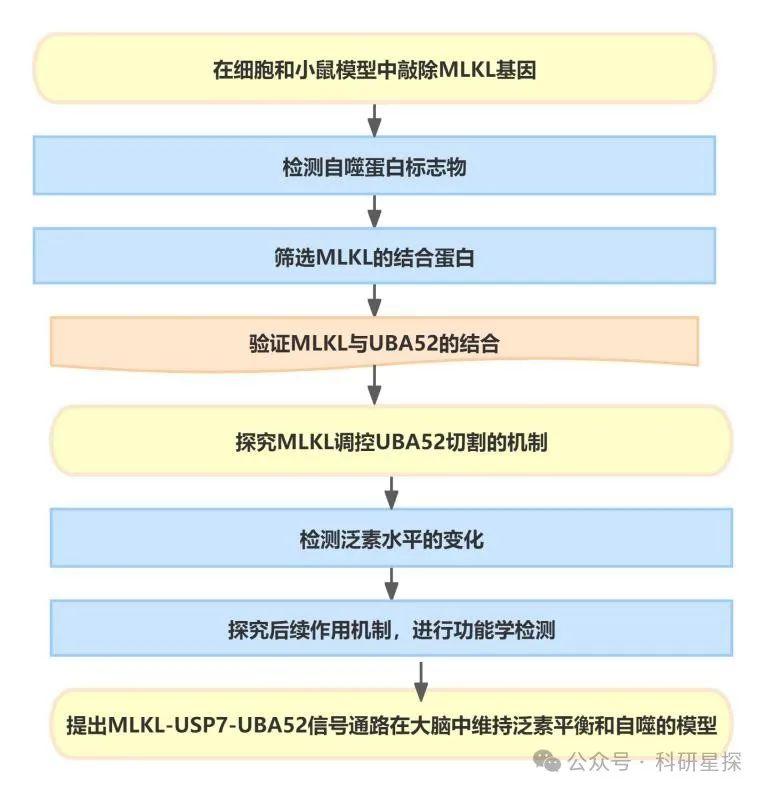
作者在神经母细胞N2a中,利用慢病毒介导的MLKL针对性shRNA(shMlkl)稳定敲低MLKL。利用免疫印迹检测自噬标志物LC3-I和LC3-II的表达水平。分别在营养条件(NR)和EBSS诱导的饥饿条件下检测。在人胚肾HEK293T细胞中用MLKL特异性抑制剂NSA处理细胞,以抑制MLKL的功能。在MLKL稳定敲低的N2a细胞中检测自噬流标志物LC3-II在自噬抑制剂处理下的变化。在MLKL稳定敲低的HT22细胞(表达荧光自噬双标记物tfLC3)中,激光共聚焦显微镜检测自噬体和酸性自噬体的数量变化。 在小鼠脑内监测神经元自噬体数量的变化。研究发现MLKL敲低或抑制剂处理均可显著降低LC3-II的表达,表明自噬体生成减少。饥饿条件下,MLKL敲低对LC3-II的抑制效果减弱,提示并非完全抑制自噬过程。在Baf存在下,MLKL敲低仍可抑制LC3-II聚积,说明自噬流减少,而非加速自噬体降解。tfLC3试验同样显示,无论自噬体还是酸性自噬体的数量均减少。在小鼠脑内亦观察到自噬体的显著下降。综上,该图明确了缺失MLKL可抑制自噬过程,这一发现奠定了后续机制研究的基础。
图1:自噬在没有假激酶的情况下被抑制
2.假激酶缺失后,自噬起始蛋白BECN1和ULK1表达下调
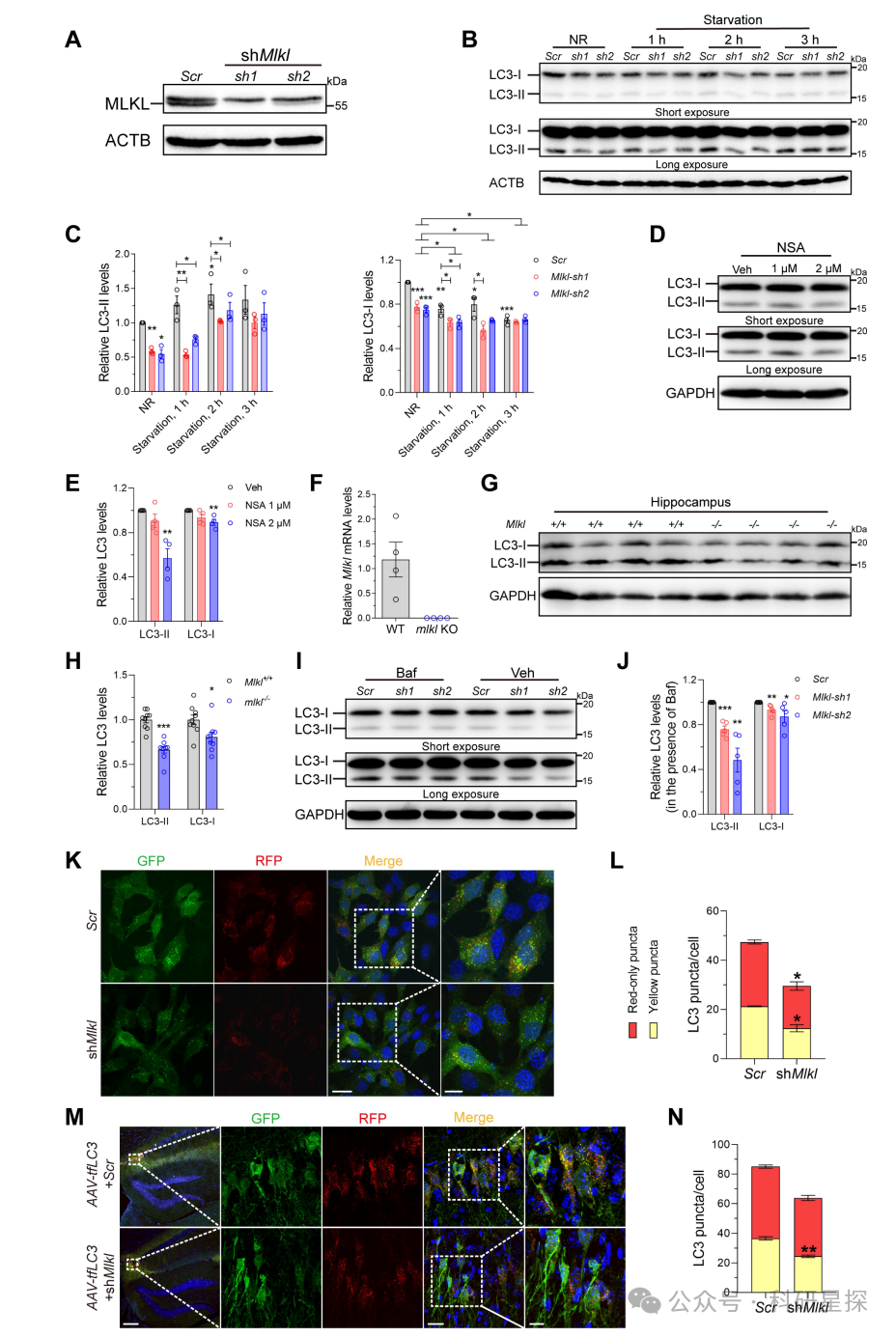
作者在mlkl敲除小鼠的海马组织中免疫印迹检测一系列自噬相关蛋白的表达。在MLKL稳定敲低的N2a细胞中免疫印迹检测BECN1和ULK1的表达。在用MLKL抑制剂NSA处理的HEK293T细胞中,免疫印迹检测BECN1和ULK1的表达。研究发现在mlkl敲除小鼠海马中自噬起始蛋白BECN1和ULK1显著下降。在细胞模型中MLKL的敲低或抑制均导致BECN1和ULK1的明显降低。但选择性基于泛素的自噬调节因子ATG14、核蛋白VPS15等未见明显变化。综上,该图明确了敲除MLKL特异性地导致两种关键自噬起始蛋白BECN1和ULK1的下调,为后续机制研究提供了线索。
图2:假激酶缺失后自噬起始蛋白BECN1和ULK1表达下调
3.泛素蛋白质组学前体UBA52鉴定为假激酶结合蛋白
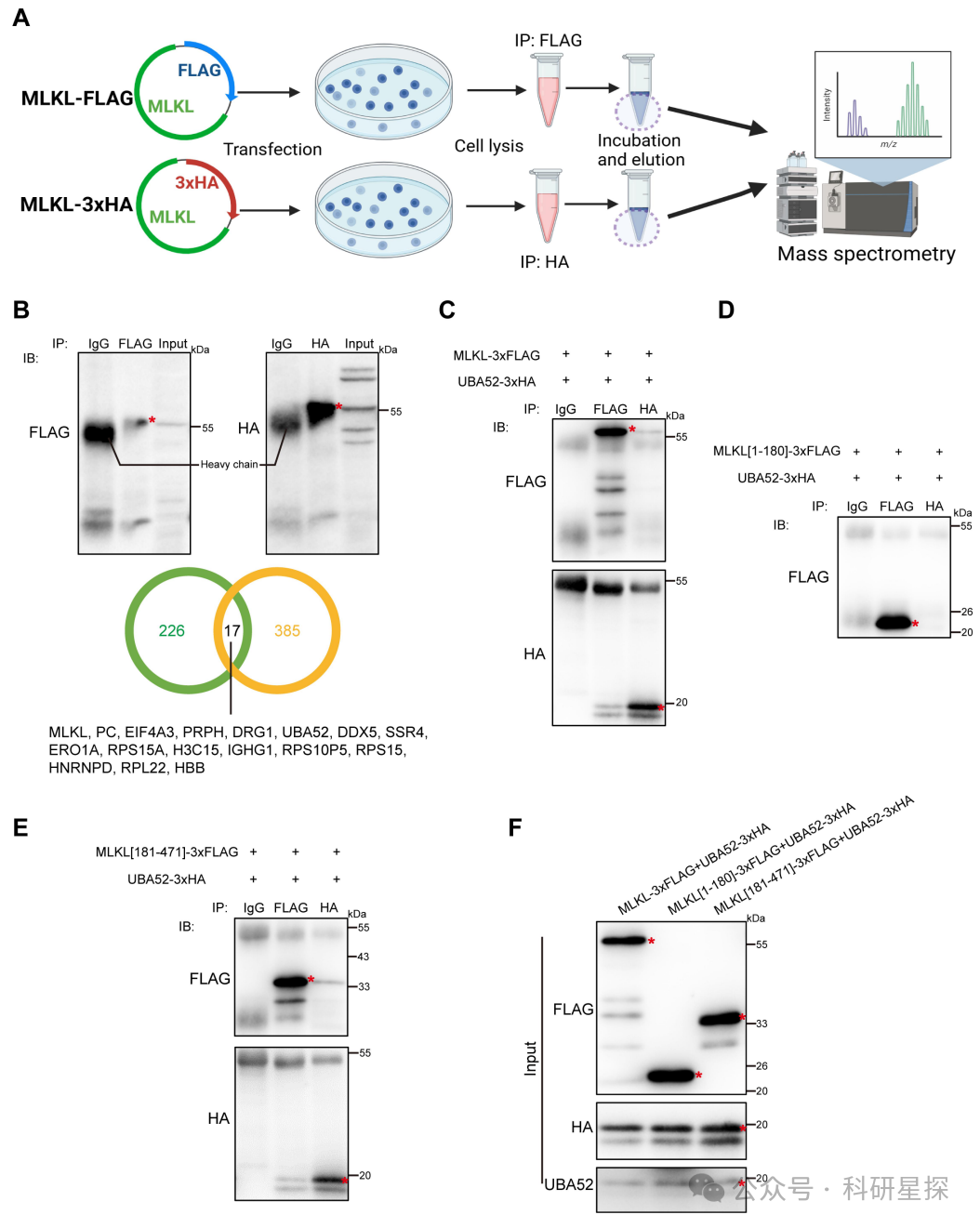
作者将MLKL的cDNA分别克隆到FLAG和HA标签的质粒中,转染HEK293T细胞后,分别利用抗FLAG和抗HA抗体免疫沉淀MLKL复合物。 SDS-PAGE分离免疫沉淀产物,考马斯蓝染色法。利用液相色谱-串联质谱(LC-MS/MS)对蛋白带进行质谱分析,鉴定MLKL的结合蛋白。根据质谱强度,筛选出与MLKL共沉淀的候选蛋白。根据亚细胞定位信息,确定泛素前体UBA52为MLKL的结合蛋白。研究发现质谱分析共检测到16个与MLKL共沉淀的蛋白。根据检测强度和定位信息,确定UBA52为MLKL的结合蛋白。后续实验验证两者可直接结合。MLKL通过激酶结构域与UBA52结合。综上,该图通过质谱鉴定了MLKL的新结合蛋白UBA52,为后续机制研究奠定了基础。
图3:泛素前体通过蛋白质组学将UBA52鉴定为MLKL结合蛋白
4.在Mlkl损失后泛素蛋白酶减少
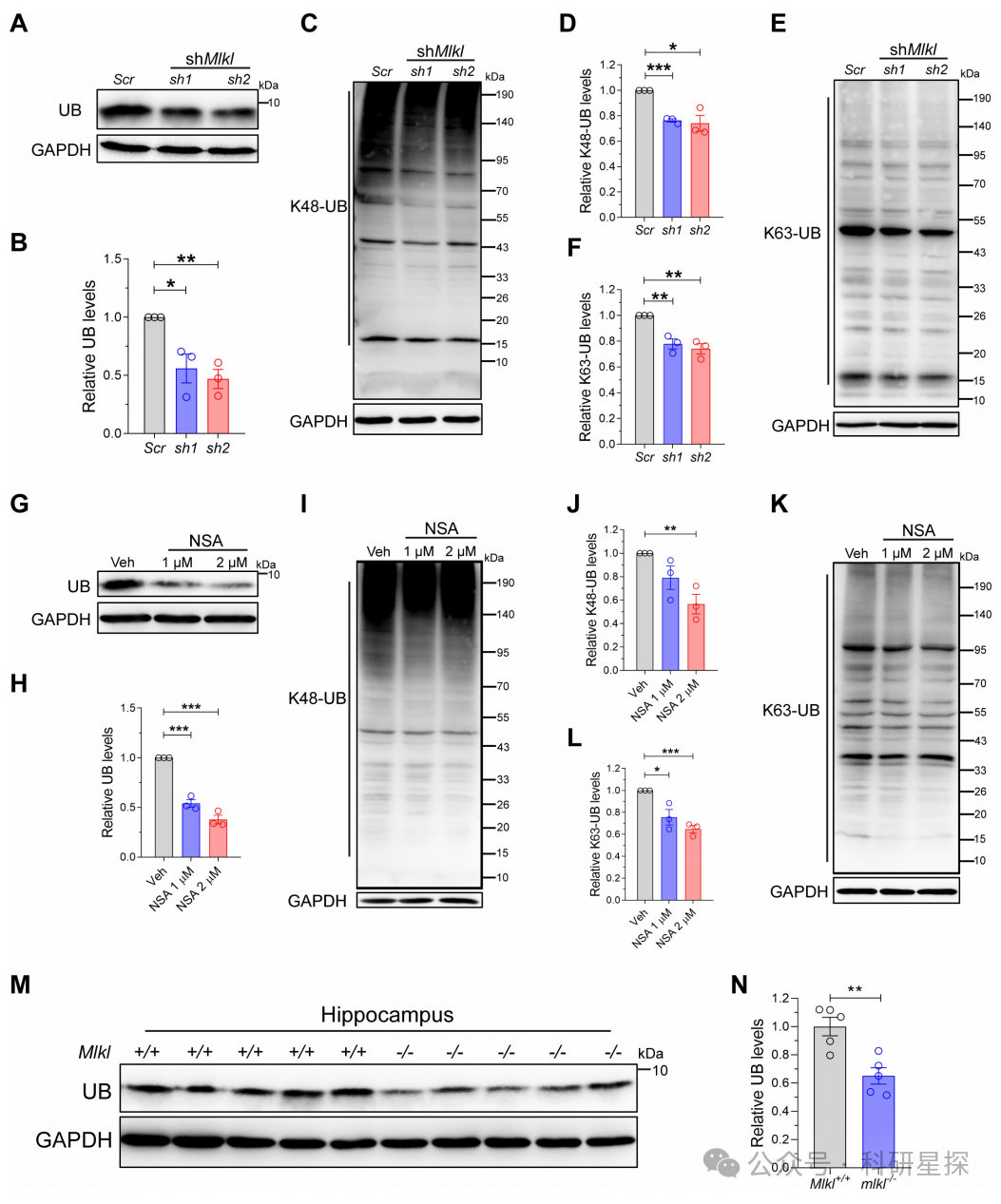
作者在MLKL稳定敲低的N2a细胞中,免疫印迹检测游离泛素(UB)以及代表蛋白降解途径的K48多泛酶化和K63多泛酶化的表达水平。在用MLKL抑制剂NSA处理的HEK293T细胞中,免疫印迹检测上述泛素种类的表达变化。mlkl敲除小鼠海马组织中免疫印迹检测游离泛素的表达水平。研究发现MLKL的敲低或抑制均可显著降低游离泛素以及K48和K63多泛酶化的水平。在mlkl敲除小鼠海马中,游离泛素减少约30%。考虑到游离泛素是大脑中优势泛素种类,其显著下降提示泛素稳态遭到破坏。综上,该图明确了缺失MLKL导致脑组织泛素缺乏,为后续机制研究提供了重要线索。
图4:Mlkl损失后泛素蛋白酶减少
5.MLKL通过大脑中的DUB USP7调节UBA52的加工
作者构建可检测UBA52全长及切割产物的质粒,转染HEK293T细胞,进行免疫印迹检测。体外合成MLKL共转染UBA52质粒入HEK293T细胞,检测UBA52全长蛋白的变化。免疫沉淀UBA52或MLKL与小鼠脑提取液,筛选可能的脱泛素化酶。体外合成脱泛素化酶与MLKL或UBA52,进行免疫共沉淀验证结合关系。研究发现敲低MLKL可导致UBA52全长蛋白堆积,产生的泛素和肽段明显减少。MLKL N端肽段是介导该过程的主要效应域。USP7被验证为参与脑组织中介导该过程的脱泛素化酶。USP7可直接与MLKL和UBA52结合。综上,该图揭示了MLKL通过调节UBA52的切割过程维持泛素稳态的分子机制。
图5:MLKL通过大脑中的DUB USP7调节UBA52的加工
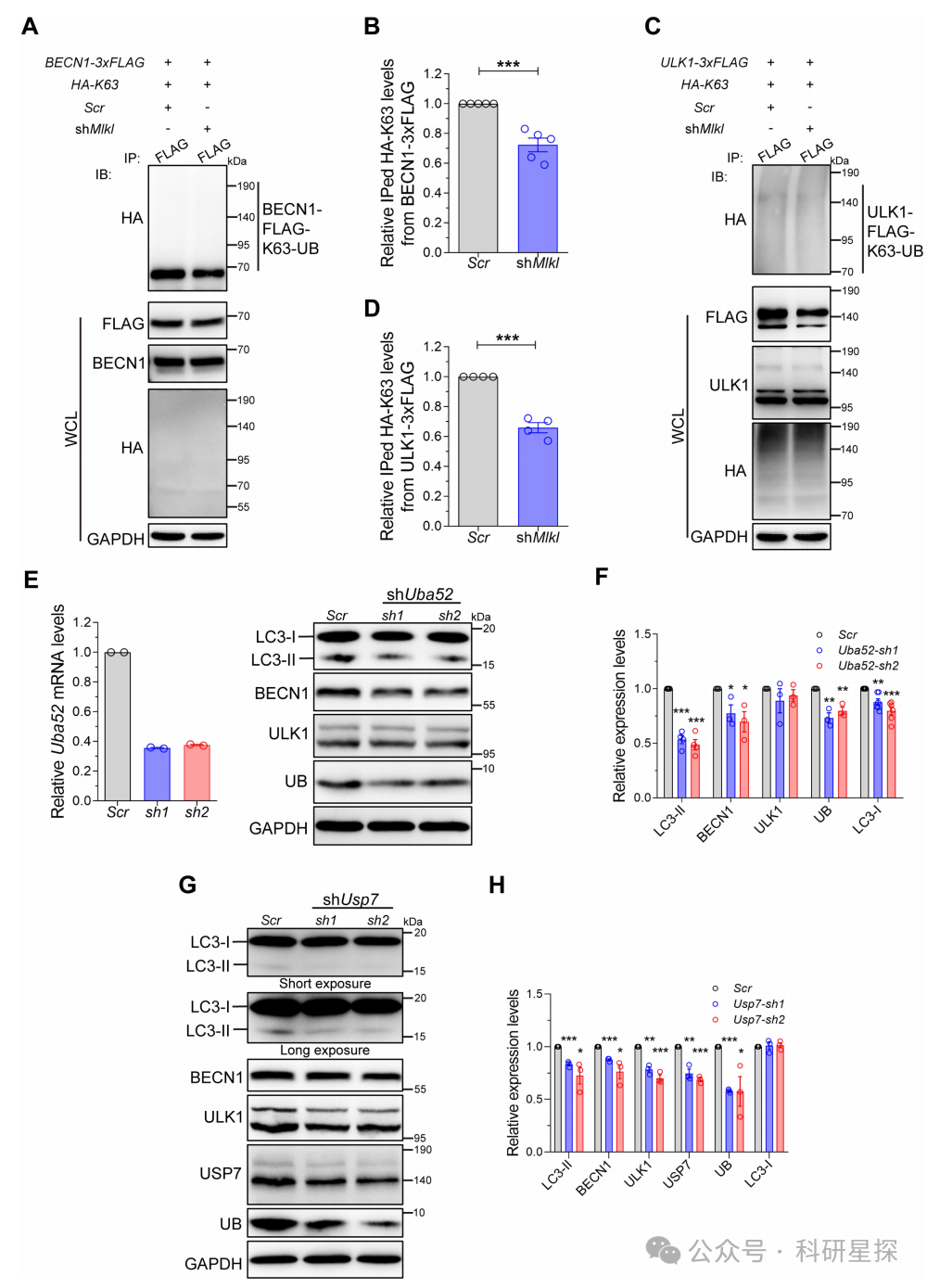
6.Mlkl缺失诱导的自噬抑制来源于泛素缺陷
作者在N2a细胞中同转染BECN1或ULK1和K63多泛酶化(HA-K63)的质粒,随后敲低MLKL,免疫沉淀BECN1或ULK1后免疫印迹检测K63的变化。在N2a细胞中敲低泛素前体蛋白Uba52和脱泛素化酶Usp7,免疫印迹检测自噬和泛素相关蛋白的变化。在敲低Uba52和Usp7的HT22细胞中,检测tfLC3报告的自噬体和酸性自噬体的变化。研究发现敲低MLKL后,BECN1和ULK1上的K63多泛酶化明显减少。敲低Uba52和Usp7同样会降低自噬蛋白的表达。tfLC3实验也显示自噬流减少。这表明MLKL缺失导致的自噬抑制源自泛素缺乏。综上,该图从泛素修改角度验证了MLKL对BECN1和ULK1表达的正向调控作用,并最终影响自噬过程。
图6:Mlkl缺失诱导的自噬抑制来源于泛素缺陷
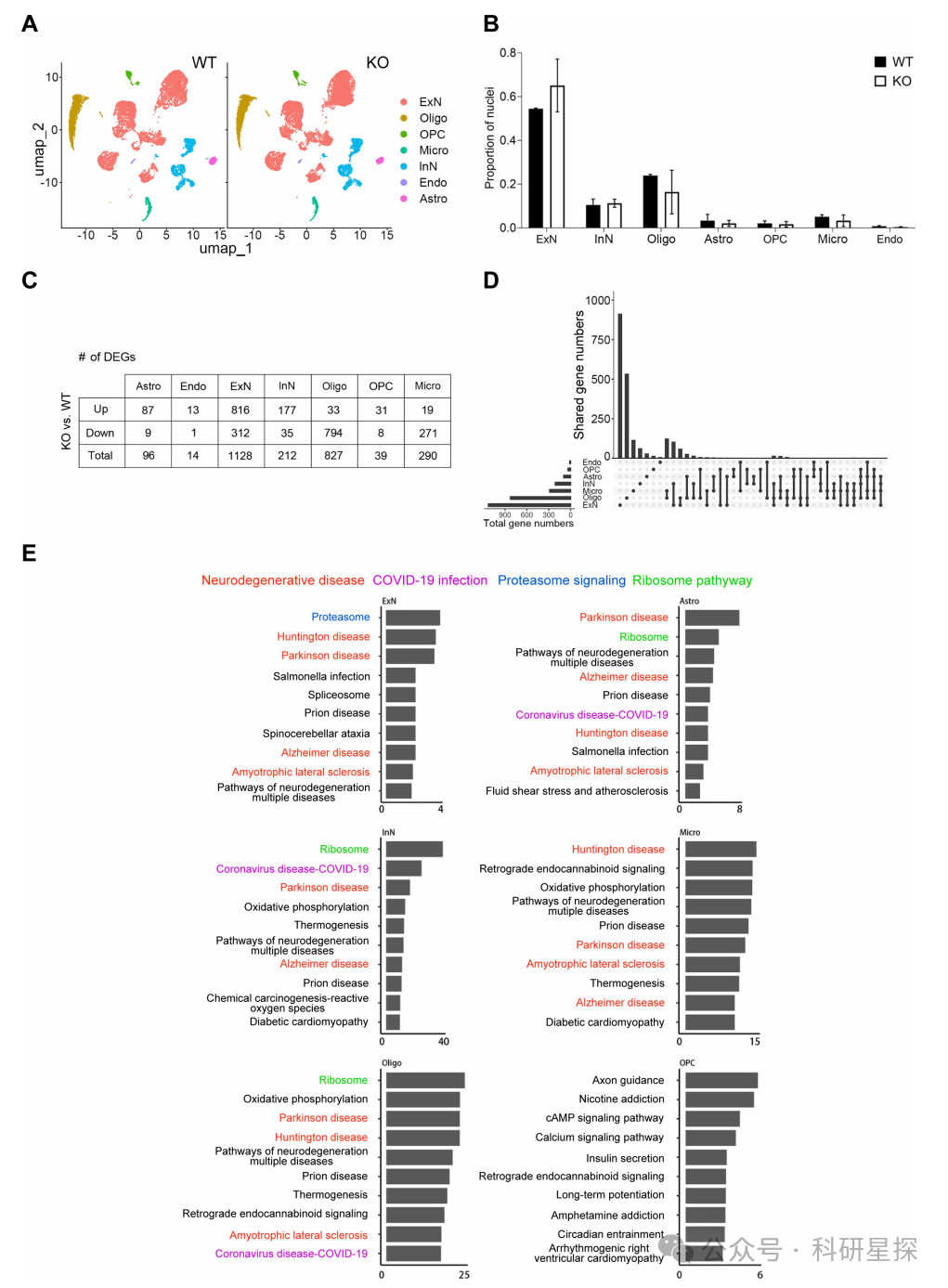
7.Mlkl缺失中断的信号通路与神经退行性疾病密切相关
作者从mlkl敲除小鼠和野生型小鼠海马中分离细胞核。利用10x Genomics的单细胞RNA测序平台进行测序。使用生物信息学分析方法进行质控,细胞聚类和差异表达基因分析。使用KEGG进行通路注释。研究发现MLKL敲除对不同细胞类型的转录组影响各异。除少突胶质前体细胞外,各细胞类型的差异表达基因均与多种神经退行性疾病相关。兴奋性神经元的通路变化与蛋白酶体途径相关,与实验结果一致。单细胞转录组结果与后续的行为学及病理学表型验证结果一致。综上,该图通过单细胞测序分析解释了MLKL敲除如何影响神经细胞,与神经退行性疾病的发生相关。
图7:Mlkl缺失中断的信号通路与神经退行性疾病密切相关
8.在AD小鼠模型中,假激酶缺失会损害认知能力,加重AD病理
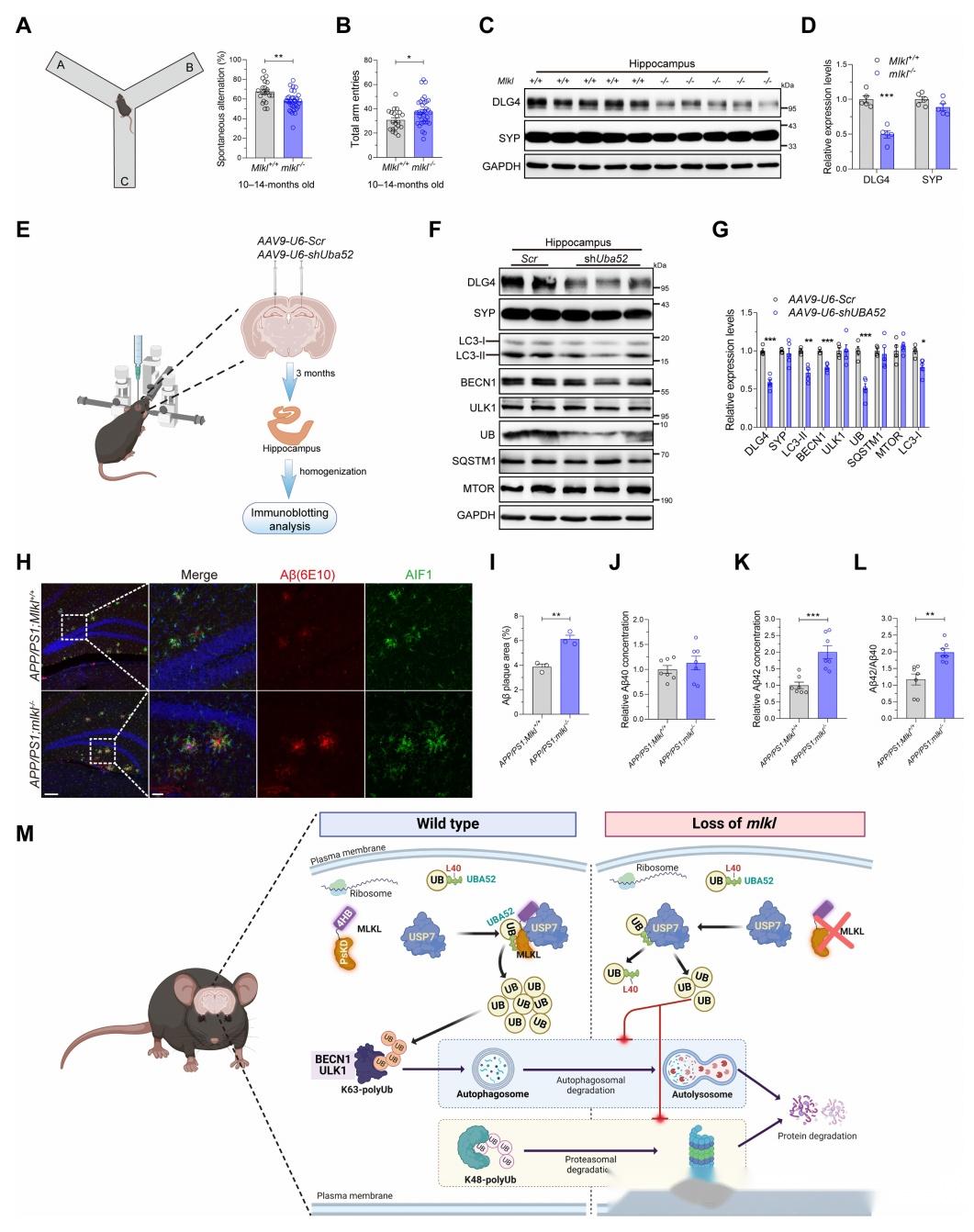
作者对mlkl敲除小鼠和野生型小鼠进行Y迷宫实验,检测空间记忆能力。免疫印迹检测mlkl敲除小鼠海马组织突触相关蛋白的变化。在野生型小鼠海马区域注射敲低Uba52的AAV,免疫印迹检测相关蛋白变化。将mlkl敲除小鼠与阿尔茨海默病模型小鼠APP/PS1交配,获得APP/PS1;mlkl+/-和APP/PS1;mlkl-/-小鼠。免疫组化和ELISA方法检测小鼠脑组织中淀粉样蛋白肽沉积。研究发现mlkl敲除中龄小鼠出现自发交替率降低,提示空间记忆能力下降。mlkl敲除小鼠突触后膜蛋白DLG4表达降低。Uba52敲低也导致DLG4下降。mlkl敲除可加重APP/PS1小鼠的淀粉样蛋白肽沉积。综上,该图验证了MLKL的缺失导致认知障碍,并可加重阿尔茨海默病病理损伤,为MLKL与神经退行性疾病的关系提供了直接证据。
图8:在AD小鼠模型中,Mlkl的缺失会损害认知能力并加重AD的病理
文章小结
这篇文章通过在小鼠和细胞模型中敲除MLKL,发现MLKL对维持正常的自噬过程至关重要。作者发现MLKL可以直接与泛素前体蛋白UBA52结合,调控其通过脱泛素化酶USP7的切割过程,从而维持脑组织中游离泛素的稳态。MLKL的缺失导致泛素水平下降,进而抑制了自噬关键蛋白BECN1和ULK1的表达。通过单细胞RNA测序分析发现,MLKL的缺失影响了与多种神经变性疾病相关的细胞通路。行为学和病理学结果验证了MLKL缺失可导致认知功能障碍,并可加重阿尔茨海默病的病理损害。本研究揭示了MLKL通过调节泛素动态平衡支持自噬过程在维持神经细胞蛋白质稳态和正常功能方面的关键作用,为防治神经退行性疾病提供了新的视角。整篇文章清晰明白,让人读起来心情舒畅,这样的文章谁能不爱呢!如果你也对这个热点和研究思路感兴趣,就来联系探长吧!
原文地址:https://zhuanlan.zhihu.com/p/17750564710
回复
使用道具
举报
提升卡
返回列表
发表回复
高级模式
B
Color
Image
Link
Quote
Code
Smilies
您需要登录后才可以回帖
登录
|
立即注册
本版积分规则
发表回复
回帖后跳转到最后一页
浏览过的版块
医学关注
标本处理
微流控技术
百科词条
病理检验
招标动态
关闭
官方推荐
/3
AI助手<小桔子>来了!
欢迎来交流,可以回答IVD行业各类问题!
查看 »
IVD业界薪资调查(月薪/税前)
长期活动,投票后可见结果!看看咱们这个行业个人的前景如何。请热爱行业的桔友们积极参与!
查看 »
小桔灯网视频号开通了!
扫描二维码,关注视频号!
查看 »
返回顶部
快速回复
返回列表
客服中心
搜索
洽谈合作
关注微信
微信扫一扫关注本站公众号
个人中心
个人中心
登录或注册
业务合作
-
投稿通道
-
友链申请
-
手机版
-
联系我们
-
免责声明
-
返回首页
Copyright © 2008-2024
小桔灯网
(https://www.iivd.net) 版权所有 All Rights Reserved.
免责声明: 本网不承担任何由内容提供商提供的信息所引起的争议和法律责任。
Powered by
Discuz!
X3.5 技术支持:
宇翼科技
浙ICP备18026348号-2
浙公网安备33010802005999号
快速回复
返回顶部
返回列表

 /3
/3 

 浙公网安备33010802005999号
浙公网安备33010802005999号 





 2026庆【网站十三周
2026庆【网站十三周 2025庆【网站十二周
2025庆【网站十二周 2024庆中秋、迎国庆
2024庆中秋、迎国庆 2024庆【网站十一周
2024庆【网站十一周 2023庆【网站十周年
2023庆【网站十周年 2022庆【网站九周年
2022庆【网站九周年

 雷达卡
雷达卡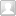 发表于 2025-3-2 07:30
发表于 2025-3-2 07:30




 提升卡
提升卡


